
Abstract
Synaptic dysfunction is a hallmark of many neurological disorders including epilepsy. An increasing number of epilepsy-causing pathogenic variants are being identified in genes encoding presynaptic proteins that affect every step of the synaptic vesicle cycle, from vesicle loading, tethering, docking, priming, calcium sensing, fusing, to recycling. These different molecular dysfunctions result in converging impairment of presynaptic neurotransmitter release, yet lead to diverse epileptic disorders. This review focuses on representative monogenic epileptic disorders caused by pathogenic variants of key presynaptic proteins involved in different stages of the synaptic vesicle cycle: SYN1 (vesicle pool regulation), STXBP1 (vesicle docking, priming, and fusion), and DNM1 (vesicle recycling). We discuss the molecular, synaptic, and circuit mechanisms of these archetypal synaptic vesicle exocytosis and endocytosis-related epilepsies and highlight the diversity and commonality of their presynaptic dysfunctions. We further discuss future avenues of research to better connect distinct presynaptic alterations to epileptogenesis and develop novel therapeutic approaches.
Keywords
Introduction
Exocytosis of synaptic vesicles loaded with neurotransmitters from the presynaptic terminal initiates electro-chemical signaling, the primary mechanism of neuronal communication in the nervous system. Presynaptic neurotransmitter release is conceptualized as a cyclic process with multiple points of regulation by thousands of proteins working in unison1,2 (Figure 1). Disruption of components of this process impairs synaptic transmission, potentially leading to epilepsy. Critical in establishing the causal connection is the discovery of over 1000 “epilepsy genes”,3,4 many of which encode presynaptic proteins. Most of these presynaptic proteins have been primarily studied for their molecular interactions and synaptic functions in reduced preparations, such as cell cultures. Therefore, it has been challenging to bridge the gap from molecular and cellular deficits to epileptic neural networks, a link that requires in-depth studies in intact brains capable of generating seizures. We propose that prioritizing a select group of key proteins from different steps of the synaptic vesicle cycle can provide critical mechanistic insights into presynaptic dysfunctions in epilepsy.
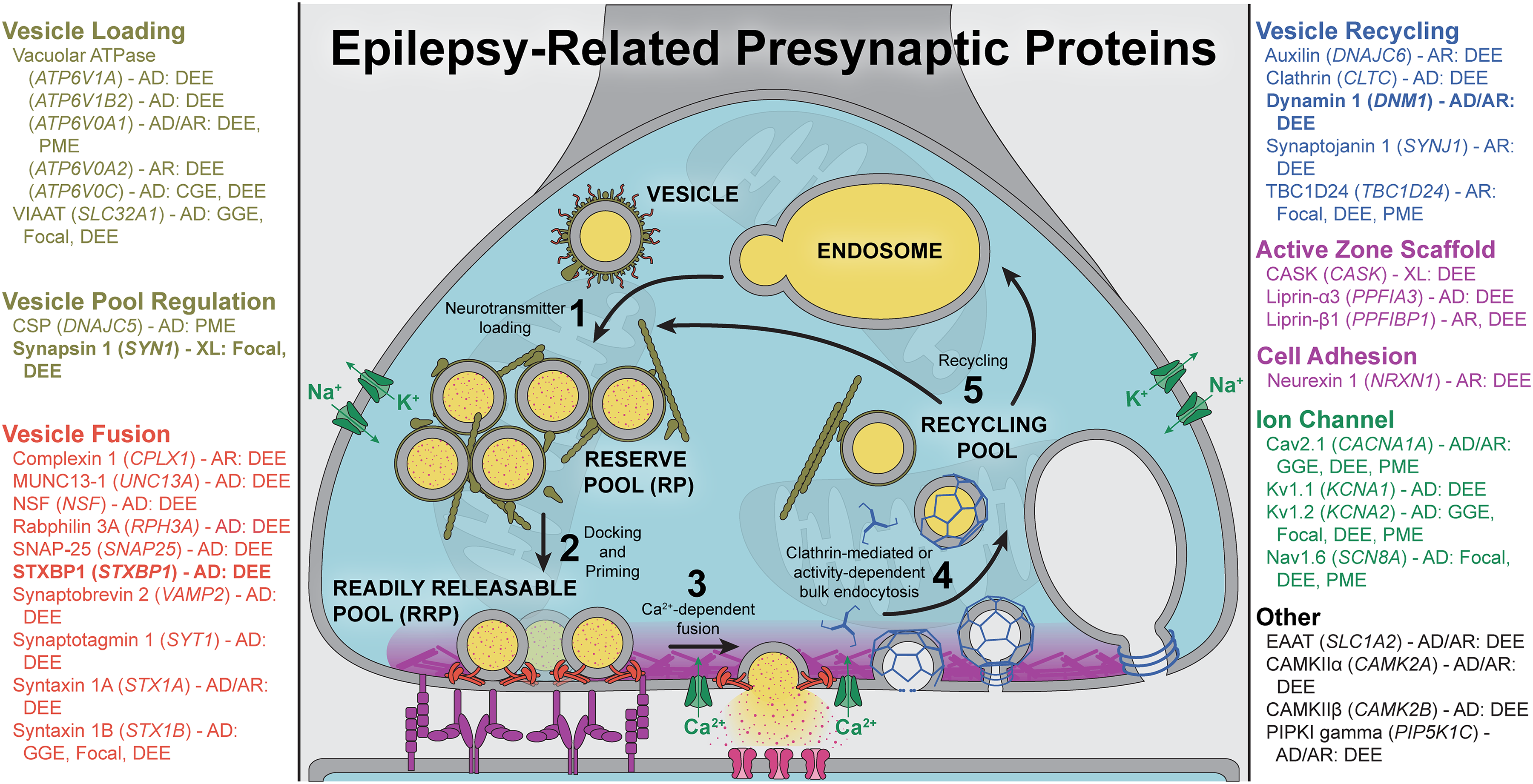
Key steps of the synaptic vesicle cycle and selected epilepsy-related presynaptic proteins. Known presynaptic proteins were cross referenced with the 2024-09 Epilepsy Gene list release from Genes4Epilepsy (https://github.com/bahlolab/genes4epilepsy). Selected epilepsy genes were additionally referenced from primary literature. Proteins are grouped and color-coded by their canonical functions and are listed by “common name (GENE) – inheritance pattern: epilepsy syndrome”. Inheritance pattern abbreviations: AD, autosomal dominant; AR, autosomal recessive; XL, X-linked. Epilepsy syndrome abbreviations: GGE, genetic generalized epilepsy; Focal, focal epilepsy; DEE, developmental and epileptic encephalopathy; PME, progressive myoclonus epilepsy; MCD, malformations of cortical development.
Advocating for this strategy, in this review we focus on the disease-relevant rodent models of selected representative monogenic epilepsies or developmental and epileptic encephalopathies (DEEs), highlighting both the diversity and commonality of their synaptic phenotypes and associated epilepsies. An important consideration when translating rodent models to human diseases, however, is the construct and face validity of the models. Many monogenic epileptic disorders are caused by loss-of-function or missense variants in a heterozygous state (autosomal dominant), whereas complete knockout of the gene or all orthologous genes are typically used to study the protein functions. Another common approach is overexpressing missense mutant proteins in a wildtype or null background. Thus, translational conclusions drawn from such studies should be interpreted with caution. Furthermore, it is often the case that the epilepsy phenotypes observed in rodent models do not cover the spectrum or severity of epilepsy in human patients. Nonetheless, rodent models provide a critical tool to connect mechanisms of presynaptic dysfunction, which are often studied in vitro or ex vivo, to network dysfunction in intact circuits that produce seizures.
Vesicle Pool Regulation: Insights into SYN1-Related Epilepsy
Tethering synaptic vesicles to actin cytoskeleton and mobilizing them from the reserve pool (RP) to readily releasable pool (RRP) (Figure 1) are regulated by synapsins (SYN) and their activity-dependent phosphorylation. 5 Truncating variants of the X-linked SYN1 gene cause familial and de novo epilepsy especially in males. A cardinal clinical manifestation is reflex seizures that are triggered by water exposure, toothbrushing, or other stimuli.6,7 Mirroring this stimulus-sensitive seizure, Syn1 knockout mice exhibit handling-induced seizures after 2 months of age,8,9 which offers a construct and face valid model to investigate how dysfunction of vesicle pool regulation may lead to epilepsy. In GABAergic neurons, Syn1 knockout reduces basal evoked release attributed to a smaller RP and RRP, which is worsened with tonic activity, indicating a dysfunction in regenerating the RRP.10–12 This deficit is specific to parvalbumin-expressing (Pv) interneurons, which have high firing rates, but not somatostatin-expressing (Sst) interneurons. 13 In glutamatergic neurons the synaptic transmission phenotypes are mild except for enhanced short-term potentiation.12,14–16 The mechanism underlying these differences is unclear, but may involve varying Syn1 levels at different synapses, leading to differences in sensitivity to its loss13,17 or distinct synapsin-dependent vesicle clustering mechanisms at glutamatergic versus GABAergic synapses. 18 Thus, presynaptic release is not a singular process across synapses, and nuance is necessary to build mechanistic understanding. It is conceivable that the reduced inhibition, which is exacerbated by intense neuronal activity due to an impairment in replenishing RRP, and the enhanced excitation can readily account for epilepsy, particularly stimulus-sensitive seizures. However, Syn1 null mice do not develop epilepsy until after adulthood despite overt synaptic dysfunction at younger ages. To further understand SYN1-related epilepsy, work remains to address how developing neural circuits become epileptic and how dysregulation of synaptic vesicle pools impacts synaptic function and network dynamics in vivo.
Vesicle Fusion Machinery: STXBP1 Encephalopathy as the Prototypical SNARE-Related Epilepsy
After synaptic vesicles are mobilized from the RP, they are docked and primed at the active zone before fusing with the plasma membrane, either spontaneously or in response to calcium influx (Figure 1). This process is orchestrated by the SNARE proteins–VAMP2, syntaxin 1, and SNAP-25–which form the SNARE complex via their SNARE motifs. Syntaxin-binding protein 1 (STXBP1, also known as MUNC18–1) and MUNC13 are essential for the proper conformational arrangements of the SNARE complex, while synaptotagmin 1 (SYT1) is the calcium sensor that triggers vesicle fusion. 19 Pathogenic variants in all these essential proteins–most of which are heterozygous–have been identified in neurodevelopmental disorders, mostly DEEs, though the prevalence and types of seizures vary widely across these conditions.20–27 Although the functions of these proteins are well understood, the synaptic and circuit mechanisms underlying these SNARE-related epilepsies remain elusive, partly due to their recent discovery and limited studies of animal models at the organismal level. One notable exception is STXBP1 encephalopathy, which is caused by heterozygous pathogenic variants and has been extensively investigated in multiple models. Importantly, STXBP1 encephalopathy is a leading cause of epilepsies and DEEs28–30 and shares many features with other SNARE-related disorders.
All STXBP1 encephalopathy individuals have intellectual disability, and 80%–90% of them have epilepsy and motor dysfunctions. 31 STXBP1 haploinsufficiency is considered as the main disease mechanism because about 50% of affected individuals carry a truncating variant. 31 Likewise, Stxbp1 heterozygous knockout (Stxbp1+/–) mouse models recapitulate key aspects of the human disorder, including epilepsy in the forms of spike-wave discharges (SWDs), the electrographic hallmark of absence seizure, and myoclonic seizures.32–34 Multiple studies of Stxbp1 haploinsufficiency reported different effects on synaptic physiology. In hippocampal cultures, both glutamatergic and GABAergic Stxbp1+/– neurons show normal basal evoked release but enhanced short-term depression. 35 In brain slices from juvenile Stxbp1+/– mice, cortical glutamatergic synapses onto striatal fast-spiking interneurons, but not those onto medium spiny neurons, show enhanced short-term depression. 33 In the somatosensory cortex, basal evoked excitatory transmission from layer 4 onto layer 2/3 pyramidal neurons and Pv interneurons is reduced, impairing feedforward inhibition and increasing excitation in this microcircuit. 36 While basal evoked GABAergic transmission from Pv interneurons onto layer 2/3 pyramidal neurons is normal at this age, 36 in brain slices from adult Stxbp1+/– mice Pv interneurons show a reduction in basal evoked release, whereas Sst interneurons show a reduction in synaptic connectivity onto pyramidal neurons. Both inhibitory synaptic connections exhibit normal short-term depression, 34 contrasting with the changes seen in hippocampal and cortico-striatal synapses. These results reveal the diverse effects of Stxbp1 haploinsufficiency across synapses and developmental stages, prompting further investigation into how these synaptic alterations may lead to different epilepsy phenotypes in adult mice.
One strategy to uncover pathogenic synaptic mechanisms is to determine the impacts of cell type-specific Stxbp1 haploinsufficiency within intact circuits, as this approach selectively alters the presynaptic outputs of targeted neurons. Notably, conditional Stxbp1 haploinsufficiency in inhibitory neurons, requiring the synergistic effects of multiple inhibitory subtypes in both forebrain and hindbrain, causes myoclonic seizures, suggesting that reduced inhibition mediates this form of epilepsy.33,37 In contrast, conditional Stxbp1 haploinsufficiency in excitatory neurons–within the cortex alone–is sufficient to generate SWDs,33,37 which was hypothesized to result from impaired recruitment of inhibitory neurons due to reduced glutamate release to subcortical structures 33 or within the cortex. 36 However, this hypothesis does not readily explain why Stxbp1 haploinsufficiency in all glutamatergic neurons fail to cause myoclonic seizures, 37 underscoring the need for further investigation of synaptic deficits in the context of neuronal circuits. Additionally, future research should determine the time course of myoclonic seizure and SWD development as well as whether their emergence correlates with synaptic alterations observed at different developmental stages33,34,36 to help elucidate the mechanisms of epileptogenesis.
Beyond truncating variants, STXBP1 encephalopathy can also be caused by missense variants with controversial mechanisms of action. Most of the experimentally studied missense variants, including the most common recurrent variant R406H, cause protein instability and a reduction of mutant STXBP1 levels rather than a gain-of-function phenotype.20,32,38–41 This aligns with a haploinsufficiency model and is further supported by investigations using neurons differentiated from patient-derived induced pluripotent stem cells carrying missense variants. 42 Some missense variants, such as R406H, can also exert a dominant-negative effect when overexpressed in Stxbp1+/– neurons by forming aggregates with wildtype Stxbp1 and reducing the functional Stxbp1 levels below haploinsufficiency.38,43 To clarify the mechanisms, additional in vivo studies, such as using knock-in mouse models, are needed to determine their effects in the context of endogenous Stxbp1 expression.
Vesicle Recycling: Closing the Gap in DNM1 Encephalopathy
After neurotransmitter release, synaptic vesicles undergo endocytosis to ensure a continuous supply of vesicles for sustained synaptic transmission (Figure 1). This process is as sophisticated as exocytosis, but fewer epilepsy-causing variants have so far been identified in endocytosis-related proteins. Among these, dynamins–a family of GTPases that catalyze membrane fission–are essential for nearly all forms of vesicle recycling. 44 Heterozygous missense variants in dynamin 1 (DNM1) have been found in individuals with DEE and are hypothesized to have a dominant-negative effect on DNM1 function.45–47 This is supported by the findings that heterozygous knock-in mice carrying missense variants R237 W or G359A display seizures,48,49 whereas heterozygous null mice do not.50,51 Furthermore, a spontaneous A408 T missense variant was discovered in the “fitful” epileptic mouse (Dnm1Ftfl). Both G359A and A408 T occur in the Middle domain and disrupt Dnm1 dimerization, which is key to its function.46,50 Though much rarer, homozygous truncating variants have been discovered in individuals with DEE who inherited them from heterozygous neurotypical parents.52,53 Thus, DNM1 encephalopathy can be modeled by both heterozygous missense variant mice (Dnm1R237W/+, Dnm1G359A/+, or Dnm1Ftfl/+) and homozygous knockout mice (Dnm1–/–), although epilepsy has not been observed yet in the latter due to early lethality.
Multiple studies in cultured neurons or brain slices from these different mouse models identified diverse synaptic phenotypes, with the caveat that the fitful mutation was studied in the homozygous state Dnm1Ftfl/Ftfl. The most consistent findings are increased synaptic depression during a high-frequency train of stimulation and slower recovery from the depression in the mutant neurons,48,50,54,55 consistent with the known function of dynamins. Basal evoked release is also reduced due to a smaller RRP, with a more pronounced effect at inhibitory synapses.49,50,54–56 This underscores defective inhibitory synaptic transmission as a potential mechanism underlying epilepsy in DNM1 encephalopathy. Supporting this notion, genetic in vivo studies reveal that Dnm1 dysfunction in inhibitory neurons led to epilepsy, with different subtypes manifesting varying severities and epileptic phenotypes.49,51 One exception is the Dnm1R237W/+ mice, where synaptic transmission in the hippocampus is enhanced at excitatory synapses and unaltered at inhibitory synapses. These mice display myoclonic jumps but not the more severe tonic-clonic seizures observed in other models. 48 It remains puzzling how these two missense variants that both have a dominant-negative effect46,48 result in such disparate synaptic and epileptic phenotypes, which also highlights the diverse effects of Dnm1 dysfunction across synapses and the need for more in vivo studies to bridge the gap between synaptic and circuit phenotypes.
Commonalities of Presynaptic Dysfunctions
By focusing on three monogenic epileptic disorders, each caused by dysfunctions of a key molecular player at one of the three main stages of the synaptic vesicle cycle, we illustrate a diverse range of disease mechanisms spanning genetic, molecular, synaptic, and circuit levels. Despite such diversity, two common themes emerge. First, presynaptic dysfunctions frequently converge on a reduced RRP, whether through impaired mobilization, docking, priming, or recycling of synaptic vesicles. Consequently, basal evoked neurotransmitter release is typically decreased. While release probability and paired-pulse facilitation or depression may vary depending on the specific proteins or mutations, sustained high-frequency stimulation generally worsens synaptic depression, reflecting a reduced capacity to maintain the RRP under prolonged activity. These deficits are likely magnified in vivo by the continuous neuronal activity in the brain. Second, studies distinguishing cell-type specific effects often reveal that inhibitory synapses are more severely affected than excitatory synapses. This may be due to differences in the molecular properties of these synapses, such as protein compositions, or variations in intrinsic physiology, like baseline firing rates. Together, these themes suggest that inhibitory synapses struggle to maintain sustained inhibition in the circuit during intense neuronal activity, explaining the paradox of how decreased neurotransmission can cause epilepsy, a disorder characterized by hyperexcitability and hypersynchrony.
Emerging Avenues of Investigation
Impaired neurotransmitter release as a critical driver of epilepsy has long been established by the discovery of presynaptic calcium channel mutations in epilepsies, 57 but ongoing exciting advances have identified epilepsy-causing variants in many presynaptic proteins. Beyond the proteins discussed above, one avenue of future research is the presynaptic scaffolding proteins that organize the release machinery at the active zone, such as CASK (calcium/calmodulin-dependent serine protein kinase) 58 and the recently discovered PPFIA3 (liprin-α3). 59 Another major research direction is to link synaptic phenotypes, traditionally studied in cultured neurons or brain slices, to circuit dynamics and dysfunctions in epilepsy, which are now readily investigated in vivo with new technologies such as genetically encoded calcium or voltage indicators, high-density silicon probes, and optogenetic tools. However, directly measuring synaptic properties during epileptic activity in vivo, likely by patch-clamp techniques, remains a significant challenge. Moreover, synaptic dysfunction occurs during development, while epileptic phenotypes often manifest later, as seen in Syn1 null mice. This temporal mismatch suggests that epilepsy may require additional epileptogenic processes triggered by synaptic dysfunction or the maturation of neural networks capable of generating seizures. Thus, gaining deeper insights into epileptic circuit formation will require developmental studies of synaptic and network properties. Lastly, monogenic epileptic disorders are promising candidates for adeno-associated virus or antisense oligonucleotide-based genetic therapies because these genetic approaches can correct protein expression and synaptic phenotypes, and validated mouse models offer robust platforms to test the efficacy of such genetic therapies in vivo, including their ability to ameliorate epilepsy.
In summary, research on monogenic epilepsy genes has advanced our understanding of how disruptions in fundamental mechanisms governing presynaptic physiology can result in epilepsy. Continued investigations into in vivo circuit deficits that link synaptic alterations to epileptogenesis will not only deepen our insights into epilepsy but also shed light on its comorbidities and an expanding list of neurological conditions linked to impaired synaptic transmission.
Footnotes
Declaration of Conflicting Interests
MX is a consultant to Capsida Biotherapeutics. Capsida Biotherapeutics provided research funds to Baylor College of Medicine to support a research project in his lab and had no role in the research, authorship, and/or publication of this article. Other authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health (F30HD114410 to KJ, R01NS100893, R01MH117089, U01NS118288, and RF1NS133657 to MX) and Texas Children's Hospital. MX is a Caroline DeLuca Scholar.