
Abstract
Developmental and epileptic encephalopathies (DEEs) can be primarily attributed to genetic causes. The genetic landscape of DEEs has been largely shaped by the rise of high-throughput sequencing, which led to the discovery of new DEE-associated genes and helped identify de novo pathogenic variants. We discuss briefly the contribution of de novo variants to DEE and also focus on alternative inheritance models that contribute to DEE. First, autosomal recessive inheritance in outbred populations may have a larger contribution than previously appreciated, accounting for up to 13% of DEEs. A small subset of genes that typically harbor de novo variants have been associated with recessive inheritance, and often these individuals have more severe clinical presentations. Additionally, pathogenic variants in X-linked genes have been identified in both affected males and females, possibly due to a lack of X-chromosome inactivation skewing. Collectively, exome sequencing has resulted in a molecular diagnosis for many individuals with DEE, but this still leaves many cases unsolved. Multiple factors contribute to the missing etiology, including nonexonic variants, mosaicism, epigenetics, and oligogenic inheritance. Here, we focus on the first 2 factors. We discuss the promises and challenges of genome sequencing, which allows for a more comprehensive analysis of the genome, including interpretation of structural and noncoding variants and also yields a high number of de novo variants for interpretation. We also consider the contribution of genetic mosaicism, both what it means for a molecular diagnosis in mosaic individuals and the important implications for genetic counseling.
The developmental and epileptic encephalopathies (DEEs) once thought to be largely due to environmental insults are now primarily attributed to genetic causes. This spectrum of rare disorders is characterized by early-onset, refractory seizures that also occur in the context of developmental regression or plateauing. 1 Individuals with these disorders have high rates of comorbid conditions, including intellectual disability (ID), autism spectrum disorder (ASD), and behavioral problems. High-throughput sequencing approaches, in particular exome sequencing (ES), have redefined the genetic landscape of this condition; still, roughly half of patients and families lack a definitive molecular diagnosis. We discuss the current genetic architecture, focusing on the last 2 to 3 years of discovery and then considering the future of genetic research in the DEEs.
Current Genetic Architecture
Some of the earliest genetic studies in the DEEs highlighted the role for de novo copy number variants (CNVs) in the pathogenesis of these disorders. Using array comparative genomic hybridization (array-CGH) techniques, up to 8% of individuals with DEE were found to carry pathogenic CNVs. 2 Similar analyses using single nucleotide polymorphism microarrays revealed pathogenic CNVs in ∼3% of individuals. 3 These CNVs, in particular microdeletions, can also be identified by analysis of read depth from ES and other high-throughput sequencing approaches. Using ES analysis, ∼3% of individuals with infantile spasms or Lennox-Gastaut syndrome (LGS) were found to have causative CNVs. 4 These CNVs can be either recurrent, as in the case of the 5q13.3 and 16p11.2 microdeletions, 2,5 or nonrecurrent.
High-throughput resequencing of candidate genes within pathogenic nonrecurrent CNVs led to some of the earliest gene discoveries, including CHD2 and SYNGAP1. 6 Since then, given the ease with which 1 to 2 de novo variants can be identified per trio exome, the majority of gene discovery has focused on the contribution of these new mutations to DEE. In general, between 30% and 50% of the DEEs can now be attributed to a pathogenic variant, the majority of which arise de novo. 7 -9 However, these estimates vary widely based on the technology used for variant detection as well as the clinical criteria used to define the cohort. For instance, epilepsy of infancy with migrating focal seizures is one of the most severe DEEs, and pathogenic variants have been identified in 69% of patients. 10 Conversely, patients with infantile spasms, characterized by early-onset spasms and hypsarrhythmia on EEG, were found to carry de novo variants in 28% of cases. 11 There are myriad genes that harbor de novo pathogenic variants, and these have been recently reviewed elsewhere 12,13 ; rather, here we focus on alternative genetic inheritance patterns that have only begun to be appreciated more recently.
The contribution of autosomal recessive (AR) inheritance to DEEs was originally thought to be exceedingly rare in outbred populations, and most early gene discoveries (eg, SLC25A22, PLCB1, and PNPO) were made in consanguineous populations. 14 -16 More recently, a small cohort study showed that ∼13% of DEE could be attributed to AR variants in an outbred population, and the majority of these variants followed compound heterozygous inheritance. 17 This contribution is likely to grow; in just the last couple of years, at least 15 new AR genes have been identified, including PLPBP, 18,19 UBA5, 20 UGP2, 21 and VARS. 22,23
There are also an increasing number of case reports of biallelic pathogenic variants in genes more commonly associated with de novo variants (Table 1). For most of these genes (CACNA1A, GRIN1, SCN1B, and KCNMA1), 25 -28,30,31,38,39 the clinical presentation for individuals with AR variants is more severe than that of individuals with de novo variants. For 2 of these genes, CACNA1A and SCN1B, increased severity is likely due to the complete absence of functional channel versus haploinsufficiency in the instance of de novo variants. 25,26,30,31 GRIN1 and SLC1A2 de novo variants are hypothesized to act in a dominant negative manner such that there is very little residual functional protein in individuals with DEE. 25,27,28,34 Thus, individuals with no functional protein (null) are only modestly more severely affected than those with de novo variants, while others with homozygous hypomorph alleles have a milder presentation of ID and ASD. Patients with de novo STXBP1 variants present with a variable DEE phenotype likely as a result of haploinsufficiency. 36 Two siblings with LGS were recently identified with a homozygous missense variant in STXBP1 that seems to act in a gain-of-function manner; the parents and heterozygous carrier sibs were unaffected. 37 Heterozygous carriers for variants in other genes (GRIN1, 28,29 SCN1B, 32 SLC1A2 34 ) were similarly unaffected. In other instances (SCN1B), carrier parents had milder epilepsies or febrile seizures. 33 These findings collectively highlight that variants can cause DEE and other epilepsies by eliciting variable effects on protein function and that recessively inherited variants can be associated with more severe clinical presentation. However, many of these instances of recessive inheritance are from single case reports, and a number of individuals were from families with consanguinity. These families tend to have large regions of homozygosity and a number of shared variants; variants falling in known genes are more likely to be reported, and this is likely true for instances of compound heterozygosity as well. For instance, a recent report of an in-frame CACNA1A 3 bp insertion was reported as a cause of progressive myoclonic epilepsy, while this is in fact a polymorphism and not at all associated with this disease. 40,41 Thus, caution is warranted in interpreting pathogenicity, and additional cases of recessive inheritance need to be identified and functional studies performed prior to these genes being considered bona fide autosomal dominant and recessive causes of DEE.
Genes With Reports of Epilepsy-Associated Variants Inherited in an Autosomal Recessive Manner and Arising De Novo.
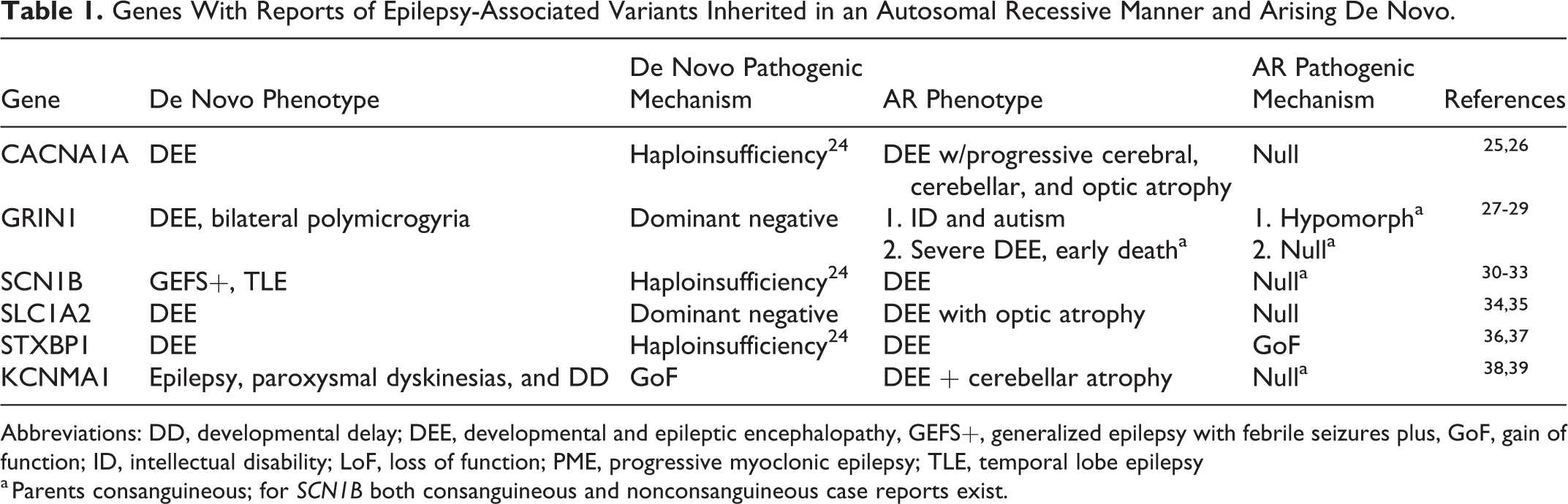
Abbreviations: DD, developmental delay; DEE, developmental and epileptic encephalopathy, GEFS+, generalized epilepsy with febrile seizures plus, GoF, gain of function; ID, intellectual disability; LoF, loss of function; PME, progressive myoclonic epilepsy; TLE, temporal lobe epilepsy
a Parents consanguineous; for SCN1B both consanguineous and nonconsanguineous case reports exist.
Pathogenic variants in the X-linked genes CDKL5, ARX, MECP2, and PCDH19 are among the most common and well-described causes of DEEs. 42 -45 Additional genes have been identified, or, in the case of X-linked ID-associated genes that are subsequently identified in individuals with DEE, reidentified. These include THOC2, 46 GABRA3, 47 and CASK. 48 The most unusual discovery, however, has been the identification of pathogenic variants in X-linked genes that affect both males and females (Table 2). For some genes (GABRA3, CASK, IQSEC2, NEXMIF), the phenotype in hemizygous males tends to be more severe than females, likely due to females having an active X with the normal allele expressing in at least a subset of cells. 47,52 -54 In females, X-chromosome inactivation (XCI) of one allele occurs to ensure equal gene dosage between the sexes. Most X-linked disorders show XCI skewing toward the mutant allele, resulting in expression of the allele that does not harbor the pathogenic variant, and thus females are unaffected. 59 However, for some DEE-associated genes, skewing is rare, and this likely contributes to clinical expression in females (Table 2). Nevertheless, where skewing was present, there was no correlation with clinical presentation. For instance, a female with a truncating NEXMIF variant had 100% skewing but no NEXMIF expression and had a clinical presentation similar to other females, 54 suggesting the normal allele was preferentially inactivated. These results should be interpreted with caution though, as XCI has been shown to be different in the brain versus the blood in individuals with Rett syndrome. 60 GABRA3 and IQSEC2 escape XCI, which may also explain the milder phenotype in females. 47,54 However, the severity of clinical presentations of females and males with WDR45 pathogenic variants is the same. This is unlikely to be due to skewed XCI, suggesting some other unknown mechanism. 57 Finally, only female patients with SMC1A pathogenic variants have been described with DEE, while male patients with pathogenic variants present with Cornelia de Lange syndrome (CdLS). Truncating variants do not lead to nonsense-mediated decay, and a dominant negative mechanism has been proposed. However, DEE in females with truncating variants is rare, and more commonly CdLS is the clinical presentation. 56 Collectively, these studies show that X-linked variants are not only important in male patients and that both sexes should be considered although severity varies on a gene-by-gene basis.
Subset of X-Linked DEE-Associated Genes Affecting Both Males and Females.
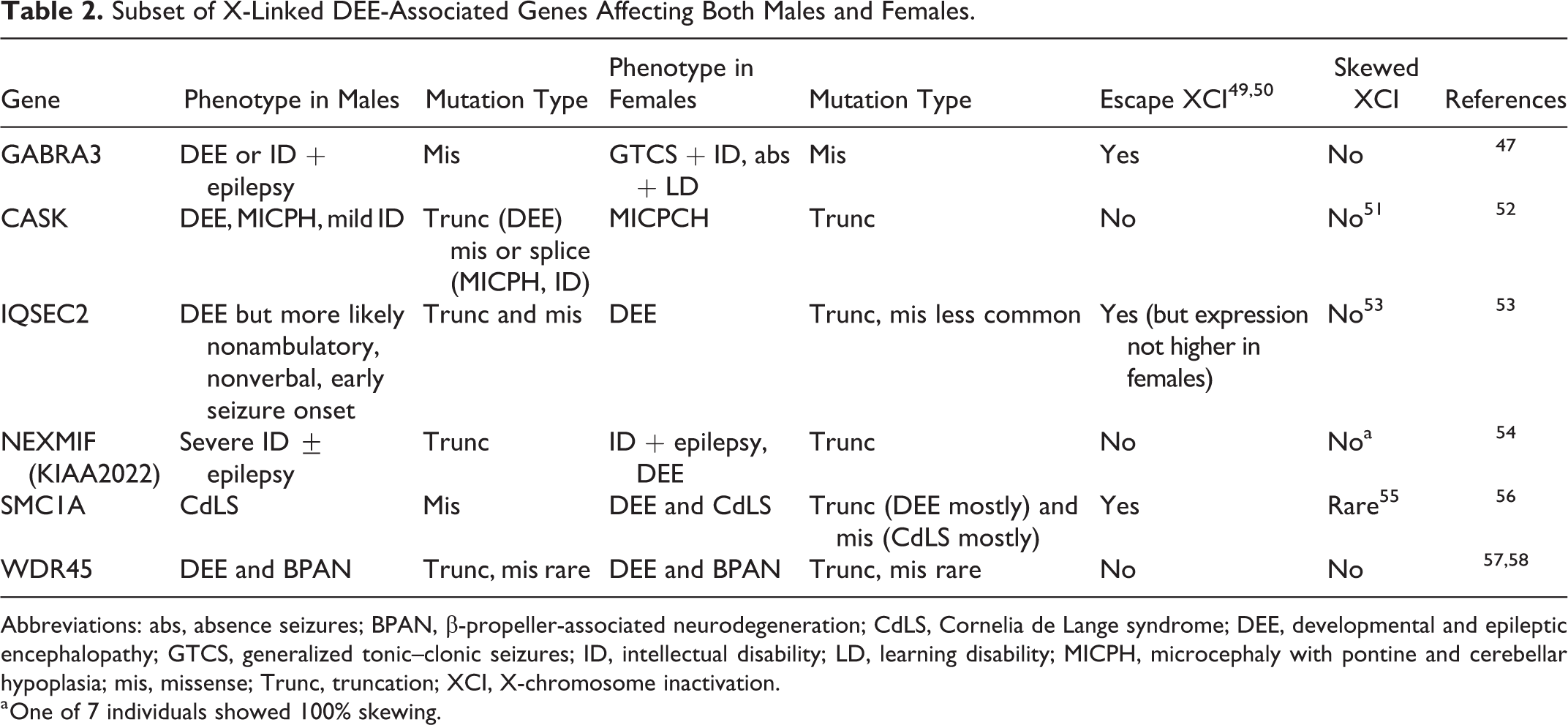
Abbreviations: abs, absence seizures; BPAN, β-propeller-associated neurodegeneration; CdLS, Cornelia de Lange syndrome; DEE, developmental and epileptic encephalopathy; GTCS, generalized tonic–clonic seizures; ID, intellectual disability; LD, learning disability; MICPH, microcephaly with pontine and cerebellar hypoplasia; mis, missense; Trunc, truncation; XCI, X-chromosome inactivation.
a One of 7 individuals showed 100% skewing.
The Future of DEE Gene Discovery
Because novel DEE genes are likely to be exceedingly rare, most gene discoveries in the recent years have been facilitated by the matchmaker exchange, the most common being Genematcher. 61 This webserver allows clinicians and researchers to enter the genetic details from individual patients into the database, and if the gene “matches,” all parties who entered in the gene automatically receive e-mails with contact details. Follow-up exchanges have facilitated the identification of novel genes, including CACNA1E, 62 CUX2, 63 KCNQ5, 64 RHOBTB2, 65 and RNF13. 66 Although this approach will likely continue to uncover new genes, the future of gene discovery lies in the hands of the commercial diagnostic testing companies who are now performing ES at a rate greater than any research endeavors.
Unsolved DEE: Beyond the Exome
Although there will likely continue to be a steady trickle of very rare novel genes discovered in DEE, most genetics research is shifting toward using genome sequencing (GS) to identify pathogenic variants in unsolved cases. Currently, short-read GS allows for the most comprehensive analysis of the genome at a feasible cost, which includes (1) better coverage of the exome (particularly in GC rich regions) and (2) detection of structural variants (SVs), including CNVs too small to be detected by array-CGH and translocations that can be detected only by karyotypes, and variants outside the exome, that is, noncoding variants that may affect gene expression or splicing. However, one of the major challenges with implementation of GS is the sheer number of variants detected; even when sequencing trios, ∼40 to 90 de novo variants are identified as compared to the 0 to 2 detected in ES. 67 -69 For this reason, most studies to date have focused solely on the exome to determine the diagnostic yield of GS. 70,71 In a DEE cohort (n = 197), the majority of whom had array-CGH and prescreening of a panel of known DEE genes, 27% of individuals had pathogenic variants (variants and CNVs) in known or novel DEE genes. 70 In a small GS study of 14 individuals with early infantile DEE, all patients had pathogenic variants that encompassed known DEE genes. 71 Of note, the majority of these variants in both studies should or would have been resolved by gene panel/ES or array-CGH.
Interpretation of up to 100 de novo variants in noncoding DNA is much more challenging, primarily because of our limited understanding of function in these regions. Perhaps the most obvious place to start is intronic regions where the candidate gene can at least be reasonably inferred. Variants in these intronic regions may impact splicing or fall within previously unannotated regions of the genome. For instance, exon 5 of SCN8A is alternatively spliced, but, up until recently, only exon 5N (N-neonatal) was captured by ES, while exon 5A (A-adult) was missed, resulting in variants in exon 5A being called “intronic.” Reanalysis of data with improved annotation led to the identification of 3 SCN8A 5A pathogenic variants in individuals with DEE. 72 This finding highlights the importance of precise annotation of the genome to identify all exons. Using brain-specific deep RNA sequencing to identify novel transcripts, 191 DEE-associated genes were recently reannotated, and an additional ∼700 kb of coding sequence was added to these genes. Mapping of ClinVar variants to these transcripts reclassified 23 intronic variants as coding, and resequencing of novel SCN1A coding regions led to the identification of 2 de novo SCN1A variants. 73 A similar study of SCN1A in patients with DEE identified 5 intron 20 SCN1A variants that likely lead to aberrant inclusion of an SCN1A “poison exon” in the transcript and the premature truncation of the protein. 74 There are likely many additional instances of tissue and cell-specific transcripts, including those harboring poison exons that may be detected using deep RNA sequencing and that may aid interpretation of variants from GS.
Finally, while GS has highlighted a role for noncoding variants in promoters and enhancers (cis-regulatory elements [CREs]) that affect gene expression in other neurodevelopmental disorders, 75,76 these studies have not yet been performed in DEE. The challenges are 2-fold: (1) our knowledge of where these CREs reside in neurons/glia that are implicated in seizures is incomplete and (2) whether a single variant can disrupt expression of a gene completely is unlikely, and initial success in this regard will likely be with SVs that disrupt CREs. To address the initial challenge, chromatin capture techniques will need to be performed in both fetal and adult brain as well as potentially iPSC-derived neurons to identify epilepsy-relevant CREs in the genome.
Unsolved DEE: The Role of Genetic Mosaicism
Somatic mosaicism has been proposed as an additional mechanism that may explain unsolved DEE. Mosaicism is the result of a postzygotic variant that arises during development. This variation is inherited by subsequent cells that arise via mitotic division, resulting in a genetically distinct subset of cells. Depending on when the variant arises during development, it could be present across many tissue types or restricted to just one. Deep sequencing of surgically resected brain tissue has shown that mosaic variants present in the brain can cause neurological disorders such as focal cortical dysplasia (FCD) and hemimegalencephaly. 77 Although FCD is one of the most common causes of focal epilepsy, deep sequencing of resected brain tissue from individuals with nonlesional focal epilepsy (NLFE) also revealed SLC35A2 variants present in the resected tissue and not in the lymphocyte DNA in 3 (17%) of 18 cases. 78
In the DEEs, studies on mosaicism have been restricted to analysis of blood, cheek swabs, or saliva from unsolved cases. For instance, analysis of 237 individuals who presented with Dravet syndrome but did not have apparent pathogenic SCN1A or PCDH19 variants revealed somatic mosaic microdeletions involving SCN1A in 2 individuals. 79 Reanalysis of commercial genetic testing panels in 9 DEE genes revealed mosaicism in 3.5% of probands. 80 There are also a number of case reports, including males with PCDH19 81,82 and individuals with GRIN1 83 and SCN2A 84 mosaic pathogenic variants. The challenge in detecting somatic variants in cases of DEE is that this ideally requires brain tissue, which is not easily accessible. Due to this inaccessibility of brain tissue, the cases of somatic mosaicism-associated DEEs are likely underrepresentative of the true number of cases.
Also worth noting here is that mosaicism has important genetic counseling implications in the instances of low-level parental mosaicism, that is, when unaffected parents carry a pathogenic variant in a subset of cells, including germ cells. Using single-molecule molecular inversion probes, Myers et al examined 123 consecutively recruited trios and identified low-level mosaicism in 8.3% of parents of individuals with DEEs caused by apparent de novo variants. 85 Similarly, a study focusing on a Dravet syndrome cohort revealed that 7% to 13% of families have a parent with low-level mosaicism in SCN1A. 86 Collectively, these studies show that roughly 1 in 10 parents will be somatic mosaics for “de novo” variants, and in these instances, recurrence risk can be up to 50%. As such, many commercial diagnostic testing companies now offer “mosaic carrier testing” for parents with children with de novo variants in DEE-associated genes.
Summary and Future Directions
Although new genes, noncoding variants, and SVs, as well as mosaicism, are likely to define a subset of unsolved cases, they are unlikely to account for all. As such, new genetic discoveries will continue to be made, including perhaps epigenetic causes as well as oligogenic models of inheritance, both of which have been shown to contribute to the “missing heritability” of other genetic conditions. 87,88 In addition to finding highly penetrant variants, genetic research in DEEs in the future will begin to unravel genotype–phenotype correlations and potential variants that modify clinical presentation. Perhaps most importantly in the coming years, we will translate DEE gene discoveries into precision medicine opportunities, including antisense oligonucleotides, new or repurposed compounds, and perhaps even gene therapy for the treatment of these disorders.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: G.L.C. has a research collaborative grant with Stoke Therapeutics, this study has no relevance to this review.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.