
Abstract
The development of antiepileptogenic and disease-modifying treatments for epilepsy is a key goal of epilepsy research. Technological and scientific advances over the past two decades have seen the development of numerous therapeutic approaches, many of which show great promise in animal models. To facilitate and de-risk the translation of these promising approaches, however, rigorous preclinical testing is needed. For the present review, we discuss challenges and approaches to conduct preclinical testing of antiepileptogenic and disease-modifying treatments in animal models.
Keywords
A primary focus of preclinical epilepsy research is the development of antiepileptogenic (AEG) and disease-modifying therapies (DMTs), recognizing that despite over 100 years of epilepsy drug development, and the approval of dozens of antiseizure medications (ASMs), 1 other than surgery, we still lack treatments that can prevent or change the course of epilepsy. Paralleling efforts in fields such as Alzheimer's disease, 2 delineating strategies to optimize preclinical studies to support clinical translation of AEG and DMTs for epilepsy is a key focus of NIH/NINDS. 3 Here, we discuss some of the major considerations for conducting these studies. Readers are referred to additional sources for more in-depth review of specific topics.4,5
Antiseizure Versus AEG and Disease-Modifying Effects
It is helpful to draw the distinction between the effects of existing ASMs and aspired for AEG and DMTs. Antiseizure medications provide symptomatic control of seizures for many patients with epilepsy, but they do not alter the course of the disease or provide lasting seizure control if drug therapy is stopped. In this regard, they resemble many cold medications which briefly control symptoms but do not shorten illness duration or provide lasting benefits once the drugs wear off. AEG and DMTs, on the other hand, should delay disease progression and/or provide continued benefits even if therapies are discontinued.
Antiepileptogenic therapies could target the two components of epileptogenesis: (1) primary epileptogenesis; the transition from a brain state in which unprovoked seizures do not occur to one in which they do occur, and (2) secondary epileptogenesis, in which preexisting epilepsy becomes more severe over time. 6 Disease modification, on the other hand, is a broader concept, but can be defined as a treatment that targets underlying disease mechanisms and alters the natural course of the disease toward better outcomes. 3 In epilepsy, DMTs might produce lasting reductions in seizures, but could also alter trajectories for epilepsy comorbidities, such as slowing cognitive decline or brain atrophy.7,8 Posing challenges for basic and clinical research, AEG and DMTs are inferred concepts, requiring a variety of evidence that an AEG or disease-modifying effect has been produced.
Acute (Provoked) Seizure Models Versus Epilepsy Models
It is widely accepted that a variety of pathological conditions and toxic exposures can produce seizures in almost all healthy mammals and many nonmammalian species. Seizures, therefore, are a common “fail state” of nervous systems. Indeed, this vulnerability has been a mainstay of ASM development, as current preclinical drug testing relies on the large selection of acute seizure models in rodents. Acutely evoked seizures generated by normal brains, however, are mechanistically different from spontaneous seizures generated by epileptic brains. While the success of ASMs at controlling seizures in many patients with epilepsy indicates the presence of some common underlying mechanisms, the failure of these drugs to show AEG effects in clinical trials9–12 highlights the limitations of acute seizure models. The development of AEG and DMTs for epilepsy, therefore, requires testing in animal models of epilepsy. 13
Discriminating Antiseizure from AEG and Disease-Modifying Effects
A critical component for testing potential AEG and DMTs is study design. By controlling seizures, ASMs can produce apparent delays in epilepsy progression even though they lack AEG or disease-modifying properties. A common approach to establish whether a drug has disease-modifying effects, therefore, is to include a drug washout period in the study design. The idea is that if the treatment has only antiseizure effects, seizure incidence will quickly revert to the baseline rate, whereas DMTs will produce lasting benefits (Figure 1(a)). This is an important approach that can provide valuable insights; however, there are some limitations that are worth considering. Firstly, not all potential therapies are reversible. Cell ablation,14,15 stem cell therapies, 16 and expression of exogenous ion channels, 17 for example, are either irreversible or not easily reversible. While such therapies might produce lasting reductions in seizures, whether they provide only symptomatic control or act as DMTs may be difficult to ascertain. For example, should one consider reintroduction of GABAergic neurons which increase inhibitory tone in the brain as being disease modifying, or as localized “drug delivery” of GABA, similar to pharmacological GABA agonists? Improved comorbidities after transplant of GABAergic progenitors in animal models hints at disease-modifying rather than solely antiseizure effects, 16 but for irreversible therapies, careful longitudinal studies, with side-by-side comparisons to traditional ASMs, may be needed to ascertain whether treatments alter disease trajectories.
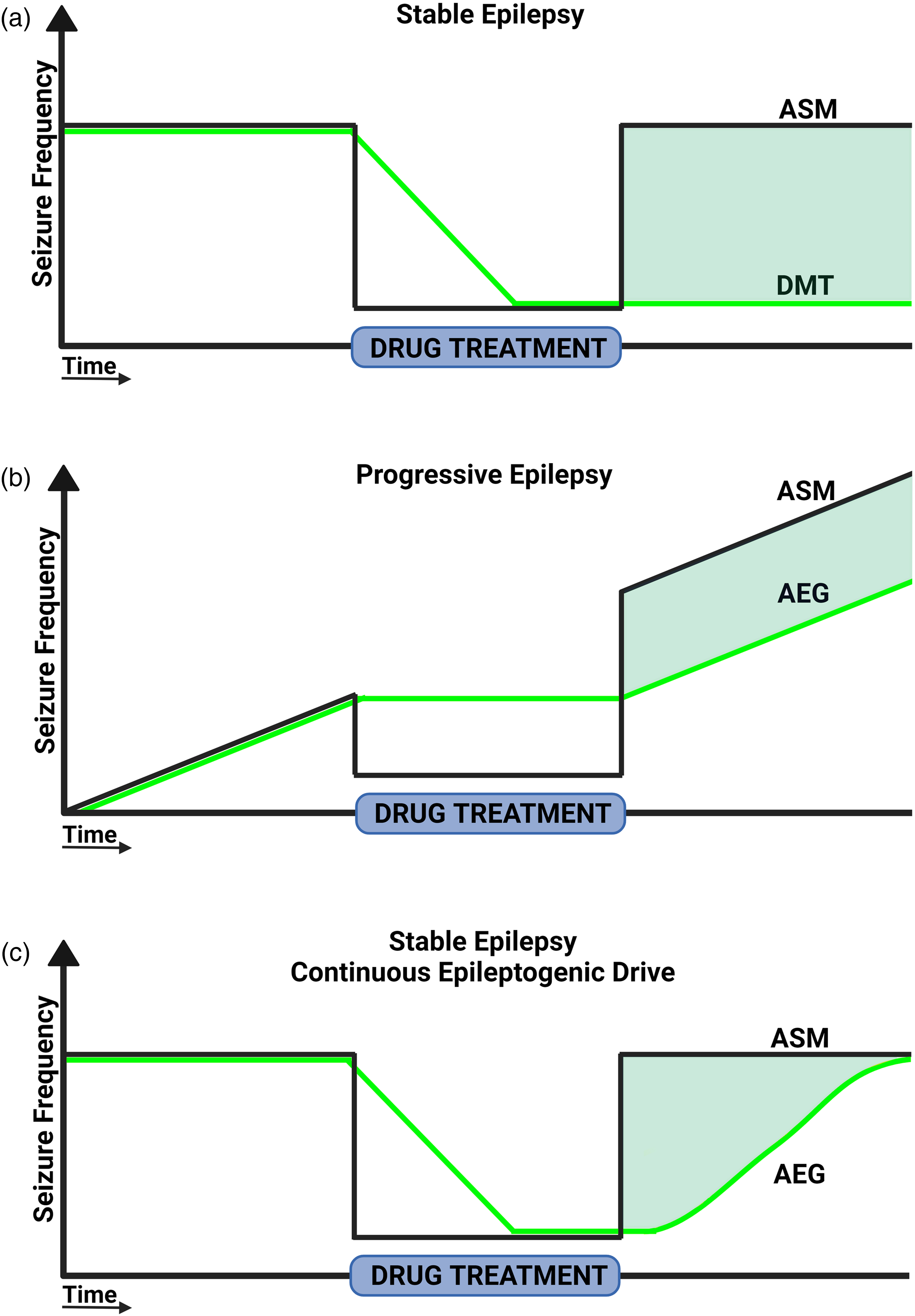
Predicted treatment responses for AEG and DMTs in stable and progressive epilepsies. Graphs model seizure frequency on the y-axis versus time on the x-axis. Predicted treatment responses to an ASM are shown in black (darker line), and AEG/DMTs are shown in green (lighter line). The bar on the x-axis denotes treatment duration. (a) ASM and AEG/DMT treatment during chronic epilepsy without secondary epileptogenesis. ASM treatment controls seizures during treatment, but seizures quickly return to the pretreatment rate upon drug removal. The DMT produces lasting seizure control even after drug removal. (b) ASM and AEG/DMT treatment during chronic epilepsy with secondary epileptogenesis. ASM treatment controls seizures during treatment, but seizures quickly return to a new, higher rate upon drug removal. The rate of progression is not changed. AEG/DMT does not reduce seizure frequency relative to pretreatment rates, but the progressive increase in seizure frequency is delayed. (c) ASM and AEG/DMT treatment during chronic epilepsy with continuous epileptogenic drive. ASM treatment controls seizures during treatment, but seizures quickly return to the pretreatment rate upon drug removal. Seizures also return to the pretreatment frequency after AEG withdrawal, but at a slower rate, reflecting a delayed reinitiation of epileptogenesis. Created in BioRender. Riley, V. (2024) BioRender.com/r09e215.
Another factor which complicates drug washout experiments and the efficacy of DMTs is the extent to which the mechanisms that drive epileptogenesis are active at the time of treatment. Although speculative, one can postulate that in some epilepsy syndromes, the mechanisms that drive epileptogenesis are transient. The transient mechanisms lead to a permanent epileptic state, but if the permanent epileptic changes can be resolved, seizures reductions persist (Figure 1(a)). Alternatively, other syndromes may exhibit ongoing epileptogenic drive, promoting secondary epileptogenesis, or reestablishing epilepsy after termination of DMTs. Ablation of newborn granule cells during the period of secondary epileptogenesis in a mouse pilocarpine model, for example, did not reduce seizure frequency but delayed the progressive increase in seizure frequency evident in controls 18 (Figure 1(b)). Persistent epileptogenic drive likely also occurs in the mTORopathies. In this disease class, which includes tuberous sclerosis complex, hemimegalencephaly, and focal cortical dysplasia, epilepsy is caused by mutations in mTOR pathway regulators. 19 Mutations produce chronic mTOR pathway hyperactivation, leading to a panoply of changes in mRNA translation, neuron metabolism, and function. 20 mTOR antagonists are effective at preventing seizures in animal models 21 and have been shown to control seizures in some patients with tuberous sclerosis.22–24 mTOR antagonists directly target the disease mechanism and thus should have disease-modifying effects; however, seizure recurrence and disease progression following drug withdrawal are evident in animal studies25–29 and patients.30,31 An obvious interpretation of these results is that despite targeting aspects of the disease mechanism (mTOR hyperactivation), treatment only restrains mTOR signaling when drug is present. Therefore, when the drug is removed, mTOR hyperactivation returns, and mutant neurons again drive seizures. Disease modification in these syndromes, therefore, might manifest as slower return to baseline seizure rates relative to ASMs rather than lasting seizure control (Figure 1(c)). This scenario is conceptually similar to established DMTs for rheumatoid arthritis, in which anti-inflammatory therapies improve outcomes, 32 but generally need to be taken lifelong, as the underlying disease-driving mechanism (autoimmune inflammation) remains despite treatment. Intermittent drug dosing has emerged as a promising alternative to continuous drug treatment for mTORopathies, with studies showing that disease symptoms can be controlled with this strategy in animal models.27,33 Careful consideration of whether and how treatments interact with ongoing epileptogenic processes is a critical component of AEG and DMT development. Although increasing costs, direct comparisons to ASMs in animal models would establish whether AEG/DMTs are actually superior and would inform the design of clinical trials.
Timing of AEG and DMTs in Acquired Epilepsy Models
Acquired epilepsy presents an intriguing scenario in which a damaging brain insult, such as traumatic brain injury, induces maladaptive changes in brain networks (i.e., epileptogenesis) to produce a new, seizure-prone brain state. This is somewhat distinct from how we think about most other neurological insults, for which treatments often focus on injury reduction (e.g., TPA for stroke). After an epileptogenic brain insult, changes occur over the course of minutes, hours, days, and years, with periods of ongoing injury and epileptogenesis likely overlapping. 5 The point at which injury stops and epileptogenesis begins is ambiguous. Effective AEG and DMTs might target injury reduction, epileptogenic changes, or both. However, as different epileptogenic processes may have vastly different temporal dynamics, it is essential that the timing of an intervention be considered when developing and testing preclinical therapies in animal models.
Broadly speaking, treatments can be initiated prior to an epileptogenic insult, after the insult but before the onset of spontaneous seizures (i.e., the latent period), or once epilepsy is established (Figure 2). Unsurprisingly, earlier interventions consistently show greater efficacy in animal models, often significantly mitigating or preventing epilepsy. It is likely that most of these interventions reduce injury severity, which naturally improves outcomes. 34 Notably, although it is possible to collect some metrics of injury severity to attempt to discriminate injury-reducing from true AEG effects with preinsult interventions, 35 the potential that interventions reduce the severity of metrics not examined is impossible to exclude, limiting interpretation. From a practical standpoint, preinsult interventions have limited translational utility, as most epileptogenic brain insults are unpredictable and thus not clinically actionable prior to occurrence.
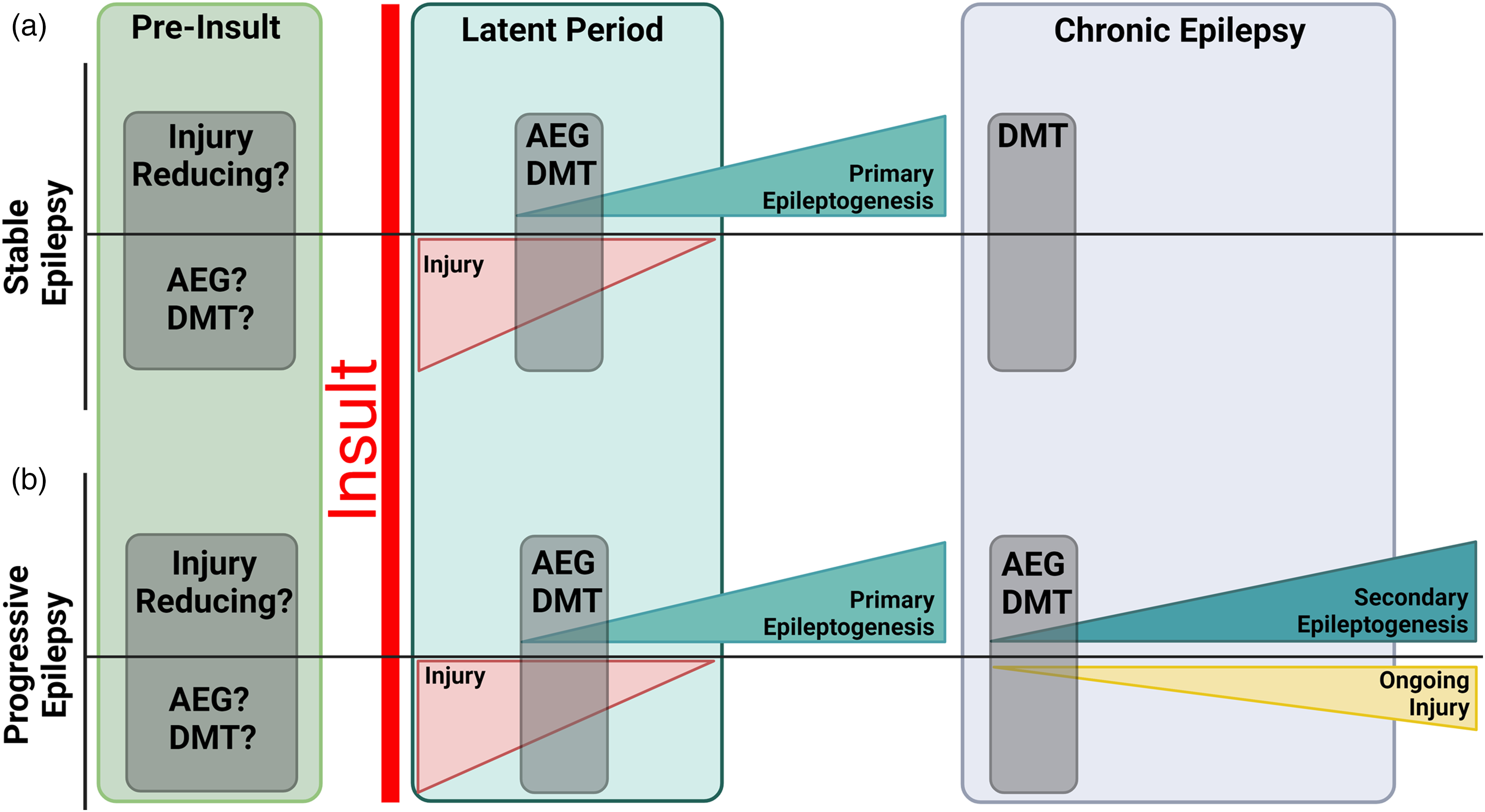
Timeline of epileptogenic processes and potential intervention points for stable (a) and progressive (b) epilepsy. In both models, the precipitating insult produces acute and subacute injury, which gradually wanes over time. The precipitating insult and subsequent injury initiate a period of primary epileptogenesis which culminates in the onset of spontaneous seizures. In stable epilepsy (a) disease progression does not occur and seizure incidence is constant over time. In progressive epilepsy, secondary epileptogenesis and ongoing brain injury occur over a protracted time period, leading to disease worsening and more frequent/severe seizures. Shaded regions highlight conceptual intervention points (1) prior to the insult, (2) during primary epileptogenesis (i.e., the latent period) and (3) during secondary epileptogenesis or chronic epilepsy. Created in BioRender. Riley, V. (2024) BioRender.com/d30s333.
Similar to therapies initiated before an insult, therapies begun during acute and subacute periods after an insult might also act primarily by injury reduction. This is because the duration of epileptogenic injury is unlikely to be rigidly constrained. Acute injury can be followed by secondary events that may produce further injury, such as spreading depolarizations, blood–brain barrier leakage, damage from inflammatory mediators, or other mechanisms. Indeed, long-term studies in animal models show progressive damage over months. 36 Preventing further injury may have AEG and disease-modifying effects, but the mechanistic distinction between therapies that act by injury reduction versus those that act by preventing epileptogenic restructuring is helpful when considering potential time windows when such agents will be maximally effective. From a practical standpoint, the hours, days, and weeks after an epileptogenic insult can be viewed as a transient period during which both neuroprotective and AEG therapies could be applied, but such therapies may need to be customized to specific injury types, etiologies, and disease timepoints (Figure 2).
Most people with epilepsy have had the disease for years; therefore, animal studies in similar chronic disease stages will have the greatest relevance to the largest population of patients. Chronic epilepsy, however, is the least studied in animal models. This is in part because studies of chronic epilepsy are time-consuming and labor-intense.37–39 In addition, chronic epilepsy almost certainly provides fewer therapeutic targets. Specifically, while decades of research reveal progressive injury and network restructuring in the days and weeks after an insult, many of these changes may stabilize over longer time courses. Changes in chronic epilepsy may also occur at a slower pace, potentially requiring protracted treatment periods to detect AEG or disease-modifying effects.24,40 On the other hand, chronic epilepsy models do offer advantages. Such models provide the opportunity for baseline EEG monitoring, which can be used to sort animals into treatment groups based on predetermined inclusion/exclusion criteria for epilepsy development and severity. Systematic randomized assignment of animals with similar seizure incidence into different treatment groups will reduce uncontrolled variability in data sets, increasing statistical power and reducing animal usage. Within animal designs, comparing baseline to treatment phases, and the opportunity for crossover approaches adds to the efficiency of studies on animals with chronic epilepsy. Finally, given the large population of patients with chronic epilepsy, preclinical testing of AEG and DMTs in chronic epilepsy models is urgently needed.
Timing of AEG and DMTs in Genetic Epilepsy Models
Status epilepticus and traumatic brain injury models are well developed and provide precise control over the precipitating insult, but lack construct validity for many types of epilepsy. Increasingly, genetic models are being developed for specific epilepsy syndromes. The traditional concept of injury followed by epileptogenesis, however, is difficult to apply in these models. This is because the precipitating event (the gene mutation) is typically present from conception, with epilepsy resulting from disruption at any point from development to adulthood. From a practical standpoint, timing interventions to disease stages when patients are typically diagnosed ensure translation utility. This may, however, miss earlier critical periods during which irreversible epileptogenic changes occur. Although mTOR antagonists show clinical efficacy in tuberous sclerosis complex, for example, animal studies demonstrate that treatment cannot reverse some defects.41,42 Basic research studies aimed at establishing key epileptogenic events in different genetic epilepsy models, therefore, need to be combined with preclinical and translational work to target these events. Like acquired epilepsy models, genetic models of epilepsy should be studied at later ages to mimic chronic epilepsy. This could provide useful data on the progression of genetic epilepsies and the development of new therapies that potentially benefit a large population of patients.
Biomarkers
Development of predictive biomarkers would greatly facilitate testing of AEG and DMTs in chronic epilepsy models, as alternative readouts could reduce the need for labor-intense long-term EEG or behavior studies while potentially illuminating aspects of disease development and progression. Although identification of biomarkers is a major focus of epilepsy research, they remain elusive.6,43,44 Dissociations between potential biomarkers and epilepsy severity are common in the literature. For example, cell death and granule cell mossy fiber sprouting, both strongly correlated with temporal lobe epilepsy, have been decoupled from epilepsy development in rodent models.45,46 Nonetheless, histological data and data on potential biomarkers are still important to collect and may become more informative as the field progresses. Studies of potential AEG and DMTs should focus on clinically relevant endpoints such as seizures and behavior. Reductions in seizure incidence and epilepsy comorbidities, combined with improved metrics for multiple putative biomarkers, provide the most compelling evidence for disease modification.
Conclusions
The development of AEG and DMTs remains a pressing need for patients at risk for and suffering from epilepsy and is a key goal for nonprofit and government funding agencies. Rapid technological advances in stem cell therapies, hIPSC generation, CRISPR technology, AAV development, designer molecules, and more have produced a wealth of promising approaches. 47 To move the field forward and de-risk clinical trials, rigorous testing in animal models is needed. Testing should focus on clinically relevant measures of disease severity, seizures, and comorbidities, but analyses of potential epilepsy biomarkers will provide additional supportive evidence, particularly when biomarkers measure putative disease mechanisms. Careful consideration of study design, appropriate animal models, and testing duration is needed to discriminate antiseizure from disease-modifying effects. No single data type or single study will be sufficient, but given the high cost and large failure rate of clinical trials, extensive preclinical testing in animal models will provide great benefits and accelerate the delivery of transformative treatments to patients.
Footnotes
Acknowledgments
Special thanks to Austin Drake for feedback on earlier versions of this manuscript. This work was supported by NIH/NINDS grants R01NS121042 and R01NS065020.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke (grant number R01NS065020, R01NS121042).