
Abstract
With more than 6000 attendees between in-person and virtual offerings, the American Epilepsy Society Meeting 2022 in Nashville, felt as busy as in prepandemic times. An ever-growing number of physicians, scientists, and allied health professionals gathered to learn a variety of topics about epilepsy. The program was carefully tailored to meet the needs of professionals with different interests and career stages. This article summarizes the different symposia presented at the meeting. Basic science lectures addressed the primary elements of seizure generation and pathophysiology of epilepsy in different disease states. Scientists congregated to learn about anti-seizure medications, mechanisms of action, and new tools to treat epilepsy including surgery and neurostimulation. Some symposia were also dedicated to discuss epilepsy comorbidities and practical issues regarding epilepsy care. An increasing number of patient advocates discussing their stories were intertwined within scientific activities.
Many smaller group sessions targeted more specific topics to encourage member participation, including Special Interest Groups, Investigator, and Skills Workshops. Special lectures included the renown Hoyer and Lombroso, an ILAE/IBE joint session, a spotlight on the impact of Dobbs v. Jackson on reproductive health in epilepsy, and a joint session with the NAEC on coding and reimbursement policies. The hot topics symposium was focused on traumatic brain injury and post-traumatic epilepsy.
A balanced collaboration with the industry allowed presentations of the latest pharmaceutical and engineering advances in satellite symposia.
Introduction
The 76th Annual Meeting of the American Epilepsy Society took place from December 2 to 6, 2022, in Nashville, TN. During this 5-day meeting, over 5500 people in-person and 680 virtually attended the meeting.
An outstanding combination of topics during major symposia, annual courses, workshops, special interest groups, poster sessions, and satellite symposia, covering epilepsy clinical and basic science, practice, and professional development, were tailored to different interests and levels of expertise. The meeting started with a symposium defining the concept of a “seizure,” which was an excellent opening to the latest science in epilepsy. Deeper knowledge about the functioning of ion channels has allowed us to target treatments in some early refractory epilepsies. Lessons from the past remind us of the wonderful observations made by Epilepsy giants such as John Hughlings Jackson. Newest pharmacotherapies, devices, and surgeries are carefully reviewed in the meeting. Special care was devoted to comorbidities, behavioral/cognitive aspects, disparities, and epilepsy care in underserved areas of the world.
The meeting is developed by members, for our members, and attempts to cater to the different constituencies of our society. An inclusive society that welcomes any other allied health professional that is involved in the care of patients with epilepsy. This includes basic scientists, neurosurgeons, advanced practice providers, neuropsychologists, nurses, and advocacy groups. The annual meeting provides guidance for clinical practice and is the perfect setting for trainees, including residents and fellows to learn about epilepsy. Patient advocates and patients are increasingly involved in the planning and delivery of the meeting.
This is a summary of the major symposia of the 2022 Annual meeting.
Epilepsy Specialist Symposium
What Is a Seizure After All?
*Barbara Jobst, MD, PhD, *Ignacio Valencia, MD, Patrick Chauvel, MD, Jean Gotman, PhD, Michael R. Sperling, MD, Jana Velíšková, MD, PhD, Yuliya Voskobiynyk, PhD
*Co-Chairs
The word seizure is derived from the Greek and means “to take hold” with the oldest scriptures going back 1000-2000 BC. Babylonian tablets have descriptions for the “Falling disease” and “to seize.” The etiology was presumed to be the effect of demons and ghosts. Around 400 BC, Hippocrates, a Greek physician, argued that epilepsy originated in the brain when an excess of phlegm (one of the 4 basic Hippocratic humors) enters the blood. The word “Epilepsy” comes from “Epi”: upon and “lepsis”: seizure. 1 Aristotelés suggested that an excess of black bile produced seizures and Galen believed that the soul was based in the brain and epileptic attacks occurred as a result of involvement of brain. The Persian physician, Avicenna, in his book, Canon of Medicine, stated that the clinical manifestation of a seizure may be associated with its origin (brain, stomach, spleen, the “Maraqq” defined as a membranous structure in the abdomen, and the whole body) or related to a specific humor. John Hughlings Jackson, an English Neurologist and father of modern epileptology stated that “A convulsion is but a symptom, and implies only that there is an occasional, an excessive and a disorderly discharge of nerve tissue on muscles.” In the 1970s, the World Health Organization and a group of experts published a “Dictionary of Epilepsy” and defined “Seizure” as a sudden and transitory abnormal phenomenon of a motor, sensory, autonomic, or psychic nature resulting from transient dysfunction due to excessive discharge of a hyperexcitable population of neurons. The International League Against Epilepsy defines an epileptic seizure as a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain. 2
This symposium aimed to provide a comprehensive concept of “seizure” from different points of view.
The long and winding road from interictal activity to seizures
Rather than defining spikes, sharp waves, or high-frequency oscillations (HFOs) as “interictal,” which implies a relationship to seizures, it may be more fruitful to consider them as pathological events that result from abnormal brain tissue, such that this abnormal brain tissue generates 3 types of events: seizures, spikes/sharp waves, and HFOs. We may then consider how these 3 event types interact with each other, how they are affected by extrinsic factors such as anti-seizure medication (ASM) or sleep, and how they can be used as a marker for each other or for the abnormal brain tissue.
An unexpected relationship links ASM and the 3 events: whereas it is often considered that a reduction in ASM, which increases seizure occurrence, is also followed by an increase in interictal spikes, it has in fact been demonstrated that reduced ASM does not result in increased spiking 3 and can result in reduced spiking. 4 The seizures (particularly those from the temporal lobe) are followed by increased spiking, which makes it look like reducing ASM is followed by increased spiking. Contrary to spikes, HFOs increase following medication reduction. 5
Sleep is another factor that affects seizures, spikes, and HFOs. Whereas the 3 stages of NREM sleep activate seizures about equally, 6 N3 is by far the state with the highest spiking rate. Here again, we see that spikes and seizures react differently to sleep.
Seizures, spikes, and HFOs are affected differently by ASM and by sleep: we should consider them as distinctive pathophysiological phenomena, which are in their specific way linked to abnormal cerebral tissue.
How are seizures defined at a microscopic level: From cell to the microcircuit to the live animal
Understanding cellular, microcircuit, and network properties underlying seizures is key to identifying effective epilepsy therapies. For example, in humans and in a mouse model of Dravet syndrome (DS) with a heterozygous loss of function of Scn1a, nonconvulsive seizures are caused by hyperexcitability of the thalamic microcircuit connected to the somatosensory cortex. Specifically, the microcircuit’s reticular thalamic cells exhibit augmented bursts of firing caused by the downregulation of calcium-activated potassium SK channels. This promotes augmented bursting of ventrobasal neurons projecting to the somatosensory cortex, where the seizures are detected. 7 Enhancing SK channel expression in the reticular thalamus or optogenetic disruption of ventrobasal neuron bursting aborts these nonconvulsive seizures in DS mice.
Thus, the thalamus both reticular and ventrobasal is required for seizure maintenance and is a promising target for DS and other genetic or acquired epilepsy disorders. 8,9
Of mice, rats, and men: what do we have in common?
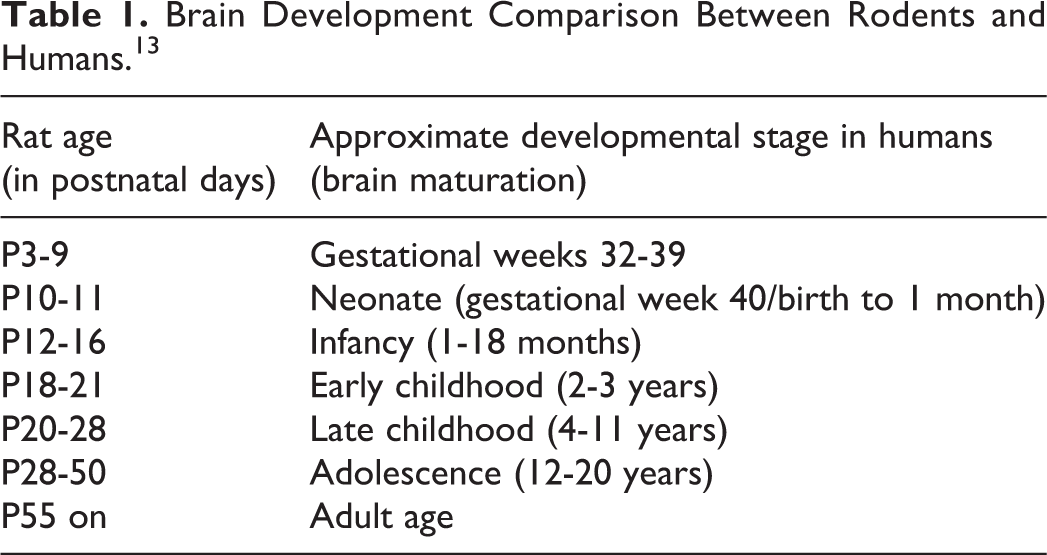
Seizure semiology in humans shares many features with rodents. Evidence from patients with acute seizures induced by convulsant neurotoxins, that is, those acting by antagonizing the GABAA receptor currents or agonists of kainic acid receptors, for example, domoic acid, show that types and progression of seizures, and histopathological consequences are similar in both species. 10,11 However, differences also exist and need to be reflected for translational interpretation of animal data. While humans develop seizures with neurotoxin doses several folds lower than rodents, rodents show higher propensity for epileptogenesis. These differences likely root from interspecies brain structural variations, especially in structures relevant to seizure generation and cessation, that is, the motor and limbic systems or basal ganglia. Proper correlation of brain developmental milestones, which occur mainly postnatally in rodents, while completed already in full-term human newborn, is also essential (Table 1). Further, the disproportional timeline for achievement of distinct brain developmental milestones in rodents and humans must be considered. 12 In conclusion, to get most of the animal models, the structural, molecular, and developmental differences between humans and rodents must be implemented when interpreting the data.
Brain Development Comparison Between Rodents and Humans. 13
Lessons learned from SEEG
Stereoelectroencephalography (SEEG) gives access to the network structure of the seizures. “Focal” seizures develop in cortical and subcortical systems; epileptogenic zone features vary depending on the system and on the underlying pathology. Mesial temporal seizures have long been considered a model. Typically, multiple limbic areas or nuclei (amygdala, anterior and posterior hippocampus, entorhinal cortex, etc.) engage synchronously. In the epileptogenic zone, the transition from interictal to ictal discharge is made up of preictal spiking, then high-frequency activity (HFA), with early and late propagation (“fingerprint” of the epileptogenic zone). Narrow-band HFA is a hallmark of this pattern. It is a tunable oscillatory phenomenon emerging from the preictal spikes. Duration and level of this narrow-band HFA are the differentiating features between the cortical systems’ seizure types. Its “chirping” aspect is characteristic of a gamma resonant activity. A new hypothesis postulates that this cortical resonance is produced by a positive plus negative feedback between pyramidal neurons and perisomatic fast-inhibitory interneurons in the epileptogenic cortex. A postinhibitory rebound would trigger the late propagation part of the seizure.
Clinical controversies in defining seizures
The International League Against Epilepsy defines a seizure as “a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain.” 2 However, this definition falls short in 2 ways: It does not include electrophysiological discharges that appear to be seizures, yet does include interictal EEG discharges that fulfill the criteria stated above. Subclinical seizures are well-described phenomena exhibiting characteristic EEG patterns identical to those associated with seizures (Figure 1) yet do not produce obvious signs or symptoms. Their electrophysiological features clearly merit a definition of seizure. At the opposite end of the spectrum, interictal spikes and even high-frequency oscillations (HFOs) may cause neurological signs, for example, by disrupting cognitive processing, 14 and should fit the definition of a seizure. However, the electrophysiological characteristics of interictal spikes and HFOs differ substantially from that of a seizure; it represents an entirely different type of hypersynchronous neuronal discharge and has different clinical implications. Lastly, the term “seizure” is overly broad and encompasses a wide range of phenomena that have vastly different implications regarding mortality, morbidity, functional impairment, and need for treatment, and the adjectives employed to classify seizures do not lessen the negative psychosocial and medical impact of a seizure diagnosis. Both the definition and terminology require updating and modification.
American Epilepsy Society-Child Neurology Foundation Symposium
Genetic Testing in Epilepsy: Improving Outcomes and Informing Gaps in Research
*Sarah A. Kelley, MD, *Anup D. Patel, MD, Katie Hentges, Leah S. Myers, Heather Mefford, MD, PhD, John J. Millichap, MD, Tamara Reynolds, MS, CGC, Jacy L. Wagnon, PhD
*Co-Chairs
The Child Neurology Foundation (CNF) connects partners from all areas of the child neurology community so those navigating the journey of disease diagnosis, management, and care have the ongoing support from those dedicated to treatments and cures. The mission of CNF is to serve as a collaborative center of education, resources, and support for children and their families living with neurologic conditions, and to facilitate connection with medical professionals who care for them. 15
Child Neurology Foundation sent a survey through the American Epilepsy and Child Neurology Society listservs and their newsletter, and all social media supported by 53 advocacy organizations and the Child Neurology Society to child neurology providers and caregivers. In total, 152 neurology providers from 30 states responded in addition to 1513 caregivers from 48 states. Child neurology providers reported 20% of their epilepsy patients did not have an underlying cause identified. Eighty-eight percent report talking about genetic testing to patients without a known cause. Among families, 40% did not know the reason for the child’s epilepsy or seizures, and among those children one-third had not had genetic testing. Interestingly, 72% of families without a diagnosis are interested in getting genetic testing yet only 35% have talked to their neurologist about it. Of those that talked about it, 32% reported their child’s neurology provider was unable to answer all their questions about genetic testing.
A standard for when to consider genetic testing is lacking. There is a need in our community to dive deeper into identifying the causes of epilepsy. Families and clinicians need to effectively communicate about options for finding the cause. CNF provided a symposium to address these issues. These included helping to determine when a genetic test is appropriate for a patient, when to order or refer a patient for genetic testing, and to go beyond seizure management to further explore a diagnosis that may subsequently affect treatment.
Improving the patient and caregiver experience
Leah Myers is the founder and Executive Director of the FamilieSCN2A Foundation, an international advocacy organization partnering with clinicians and industry to cure the life-limiting conditions caused by mutations of the SCN2A gene. She’s also the parent of 12-year-old Ben, whose diagnosis a decade ago launched her family’s own heartbreaking and miraculous journey through genetic epilepsy. Finding the etiology was just the start of her fight that has had an incredible ripple effect. The Foundation represents more than 1000 families globally, funding research, and supporting families. Leah hopes that by sharing her story she will inspire professionals to work together with family-driven organizations. Just like no 2 patients are the same, no 2 parents are the same. Do not shield the families—empower them, encourage them to learn, and become experts. Because no one will fight for a better future for pediatric genetic epilepsy like parents trying to save their child.
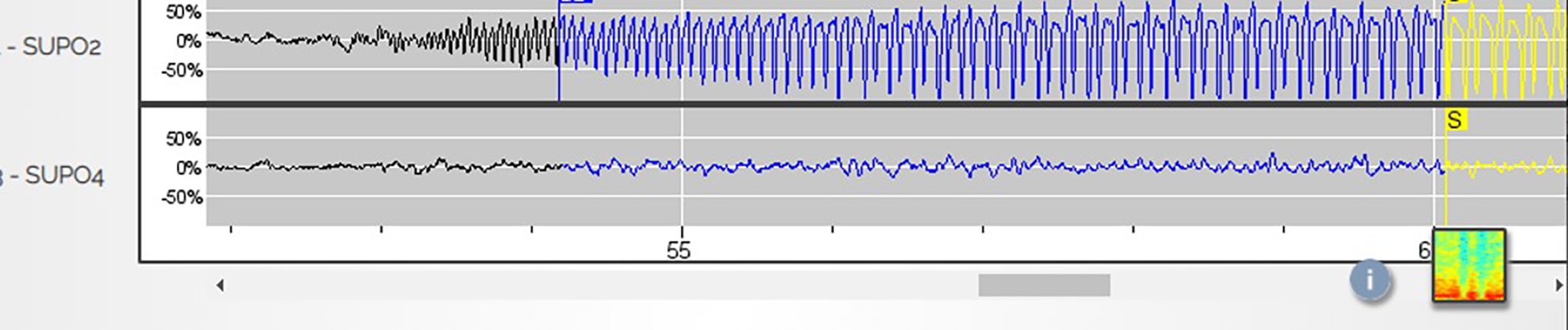
Subclinical seizure on a depth recording.
Impacts on clinical care
The number of genes associated with epilepsy has increased dramatically over the last 30 years, resulting in an increasing clinical impact of genetic testing on the diagnosis and treatment of patients with epilepsy. 16 Several established genes have known treatment implications, including SCN1A, POLG1, ALDH7A1, SLC2A1, SCN2A, and KCNQ2. KCNQ2 was one of the first known epilepsy genes discovered to cause neonatal seizures that are passed down in families (now known as “Self-limited neonatal epilepsy” [SeLNE]). 17 In 2012, different pathogenic variants in KCNQ2 were associated with severe intractable neonatal-onset encephalopathy with a variable spectrum of outcomes in seizures and development (now known as “KCNQ2-developmental and epileptic encephalopathy” [KCNQ2-DEE]). 17 Between 2012 and 2017, there was increased awareness of the severe presentation and it was determined that sodium channel-blocking anti-seizure medications are most effective for seizures in this disorder. 18 Five years later, neonates with unexplained seizures still do not receive genetic testing that could stop the diagnostic odyssey, facilitate counseling, and lead to personalized treatments. 19
Bench to bedside: How basic research informs treatment in genetic epilepsies
Functional analyses of genetic variants can help us identify molecular mechanisms underlying genetic epilepsies. In turn, details of these pathogenic mechanisms can inform treatment with available anti-seizure medications and spur development of new therapies for genetic epilepsies that are refractory to current treatments. For example, we have tools to study the properties of sodium currents generated by variants in sodium channel genes identified in DS (SCN1A) and SCN8A-related developmental and epileptic encephalopathy (DEE). 20 These studies revealed that DS is caused mostly by loss-of-function variants of SCN1A. In contrast, SCN8A-related DEE is caused by gain-of-function variants of SCN8A. These data explain why sodium channel blockers are contraindicated for Dravet syndrome but may be helpful for SCN8A-related DEE. Promising new therapeutic strategies utilizing antisense oligonucleotides are being developed that upregulate SCN1A or downregulate SCN8A to treat sodium channel-related epilepsies. 21,22 Thus, basic research can inform and impact clinical care in genetic epilepsies.
Genetic testing in epilepsy
The number of genes in which pathogenic changes can cause epilepsy has grown rapidly over the past decade. Alongside that growth, the array of genetic testing options has expanded from chromosome arrays and single-gene testing to include gene panels, exome sequencing, and genome sequencing. In addition, methylation testing and RNA sequencing are increasingly employed in specific clinical situations, often to clarify or augment prior test results. Selection of the appropriate genetic test(s) in the clinical setting should take into consideration the diagnostic yield of each test. In epilepsies, exome sequencing is most efficient, and recent guidelines from the National Society of Genetic Counselors recommend exome or genome sequencing, when possible, as the first-tier test in individuals with epilepsy; a gene panel with >25 genes can be performed if exome/genome testing is not possible, followed by chromosome array. Given the complexities or both genetic testing and interpretation of results, pre- and posttest genetic counseling should also be incorporated into the testing process. 23
Managing common genetic testing barriers
Over the past 20 years, a rapid increase in the volume and complexity of genetic testing has led to gaps in provider training, insurance coverage, and adjustments to hospital workflows and staffing, resulting in barriers to patient access. As the clinical utility of genetic testing advances, there is an increasing need for neurology provider to integrate genomics into their practice. 24 In order to ensure patient access, creating new delivery service models, collaboration between departments, and increased provider support, time, and education is needed. Short-term strategies to overcome testing barriers include a partnership with genetics and social work departments, engaging the existing resources in the commercial genetic testing lab, and utilizing existing staff in new ways. 25 Long-term strategies include creation and implementation of new workflows, increased genetics support staff, and increased provider training at both the graduate and postgraduate level. 24
Knowledge gained from genetic testing is exploding but there continue to be barriers to getting clinically relevant testing for our patients and to learning more about how genetic testing can help them. The talks during this symposium demonstrated how we are making progress toward these goals. We have learned how important it is for families to have access to genetic testing and information not only to inform their provider’s treatment decisions but to give them an opportunity to connect to advocacy groups and to other families with similar diagnoses and challenges and subsequently empower those families. With genetic testing knowledge, we can treat the whole patient and not just epilepsy in isolation. Exciting research allows us to continually learn more as studies are taken from the bench to the bedside and then back again. If we do not get an answer the first time around, we now have new emerging approaches to evaluating further and work toward finding a diagnosis for our patients. These wonderful new options require increased training and improved models to better deliver this care while expanding genetic counseling services so we can continue to improve the utility of genetic testing to the benefit of our patients and their families.
Spanish Symposium
Seizures and Use of Electroencephalography (EEG) in the ICU
*Jorge G. Burneo, MD, MSPH, Miguel Arevalo-Astrada, MD, Maite La Vega-Talbott, MD, Luis Carlos Mayor, MD, Clio Rubiños, MD, Cesar Santana-Gomez, MsC, PhD
*Chair
The role of high-frequency oscillations in epilepsy, epileptogenesis, and drug-resistant epilepsy
High-frequency oscillations (HFOs) are defined as local field potentials corresponding to an increase in bursts of synchronous neuronal spikes. Based on their spectral frequency properties, HFOs are classified into “ripples” (80-200 Hz) and “fast ripples” (FRs, 200-800 Hz). Experimental findings from animal models and brain tissue from patients with drug-resistant epilepsy (DRE) showed the association of the HFOs with epileptic tissue and, are increased in areas of the brain capable of generating epileptic seizures. In clinical epilepsy, recording HFOs could provide a measure of the risk for developing epilepsy and propensity for spontaneous seizures, and, in some types of epilepsy, the epileptogenicity of a lesion and severity of the disease.
Rhythmic and periodic patterns in critically ill patients
In critically ill patients, the EEG may reveal interictal discharges but also rhythmic and periodic patterns (ictal–interictal continuum), as well as ictal (seizures and status epilepticus) and potentially ictal activity (BIRDs).
Rhythmic and periodic patterns consist of the repetition of a waveform at nearly regular intervals and are described according to the localization, type of pattern, and presence of modifiers, which helps to predict the risk of seizures. 26 They constitute the interictal–ictal continuum when they do not meet the criteria for ictal activity.
A seizure is defined by the frequency of the discharges, duration, clinical manifestations, or response to parenteral anti-seizure medication. Status epilepticus is a seizure of longer duration (≥10 continuous minutes or a total duration of >20% of any 60-minute period of recording) and BIRDs are not seizures because of their short duration (<10 seconds).
The use of continuous EEG in the critical care setting: What do we do when resources are limited?
Video-EEG allows us to identify nonconvulsive seizures and nonconvulsive status epilepticus. It is also useful for the assessment of those affected by abnormal mental status following generalized status (SE) or a seizure. Other indications would include the comatose patient, acute supratentorial brain injury with altered mental status, unexplained altered mental status, and assessment of paroxysmal events. 27
In locations with limited resources, medical history, mental status, and comorbidities may help with the diagnosis. But, in most situations the use of cellphone videos is useful. If the patient is hospitalized, a 1-hour EEG would suffice. If the answer is not met, repetitive studies or the use of video-EEG for 6 hours may help. 28
The most common findings include periodic discharges, focal or generalized, abnormal background, and lack of background reactivity. The presence of epileptiform discharges in the first 30 minutes of recording, increases the risk of status epilepticus. If there is no presence of epileptiform discharges in the first 2 hours of recording, the risk for SE decreases to 5%.
Case illustration of seizures in a critically ill patient
During this talk, we consolidated the audience’s knowledge of the latest American Clinical of Neurophysiology Society ICU-EEG terminology, 26 including new terminologies such as Ictal interictal continuum (IIC), possible electrographic seizures, and cyclic alternative pattern of encephalopathy (CAPE). We reviewed and analyzed frequent patterns seen in critically ill patients by promoting an interactive discussion focused on the clinical implication and possible management of the patterns aiming to provide precision medicine management for each case. We taught the audience the concept of benzodiazepine/anti-seizure medication trials and discussed cases when the trial’s results were positive, negative, or equivocal. 29 Finally, we reviewed the importance of using quantitative EEG and video as adjunctive diagnostic tools for continuous EEG monitoring.
Annual Fundamentals Symposium
Beyond Seizures: Tapping Into the Community for Care
*Anne T. Berg, PhD, *Martha Sajatovic, MD, Barbara Jobst, MD, PhD, James W. Mitchell, MBChB, Janelle L. Wagner, PhD
*Co-Chairs
Seizures are the core defining symptom of epilepsy and the focus of most treatment efforts. While critical to the definition of epilepsy, seizures alone are not the only aspect of having epilepsy and often are not the most burdensome aspect of it. Many other aspects and consequences of epilepsy can have a profound impact on health-related quality of life and on the success of people with epilepsy but are often not the direct subject of clinical care. This symposium addressed (1) key areas of nonseizure outcomes identified as important to patients by patients and caregivers, (2) their recognition and their treatment implications, and importantly, (3) the role of self-efficacy, self-management, and partnerships with services outside of the epilepsy care setting to optimize these outcomes and ultimately the overall care and quality of life for people with epilepsy and their families.
International epilepsy standard set of outcomes for routine clinical practice
The International Consortium for Health Outcomes Measurement (ICHOM) 30 has developed an international Core Outcome Set for routine clinical practice for infants, children, and adults with epilepsy, which can be used for comparisons across countries and settings as well as quality improvement within a setting. Delphi-based consensus methods engaged an international working group of health care providers, epilepsy researchers, people with epilepsy, and their representatives to identify a set of 22 essential outcomes. Brief and feasible to implement measurement tools including Patient Reported Outcome Measures (PROMS) were recommended based on their evidence of strong clinical measurement properties and cross-cultural applicability. The essential outcomes included many nonseizure outcomes: anxiety, depression, suicidality, memory and attention, sleep quality, somnolence, and—for infants and young children—neurodevelopmental outcomes.
The ICHOM Epilepsy Sets ensure that outcomes that are relevant to people with epilepsy, their representatives, and health care providers are measured. They also facilitate harmonization of outcome measurement, and if widely implemented should reduce outcome measurement heterogeneity and therefore facilitate comparative research and big-data science. 31,32
Behavioral health across the spectrum of epilepsy
Behavioral health comorbidities are common in persons with epilepsy (PWE), can worsen ASM side effects, and are associated with poor health-related quality of life (HRQoL). 33 In addition, 30% to 60% of PWE are nonadherent to ASMs. Results from a recent scoping review revealed that socioeconomic factors influenced adherence, educational, seizure, and HRQoL outcomes in youth with epilepsy.
Addressing behavioral health comorbidities can reduce the seizure burden and improve quality of life. Evidence-based screening measures are available and have clinical utility (e.g., brevity, no cost). 34 A 3-tiered health promotion approach provides more intensive services for patients at higher risk. 35 In applying this stepped-up model to pediatric epilepsy care, providers are encouraged to consider the family background and social determinants of health when setting up a behavioral health screening protocol for (1) a targeted population (high-risk, new patients), (2) personnel (clinic nurse, behavioral health), (3) method (survey, waiting room), and (4) next steps in care (documentation, positive screen). 34 Integrated behavioral health care reduces challenges with access and stigma, increases patient satisfaction and communication between providers, and embodies comprehensive epilepsy care.
Addressing the cognitive difficulties of people with epilepsy
Cognitive difficulties are common in people with epilepsy and interfere with quality of life. Impaired cognition often contributes more to disability than the seizures itself. Cognitive difficulties in epilepsy have many etiologies and interictal epileptiform activity has been shown to selectively interfere with cognitive processing. Treating epileptiform activity with ASM is problematic as medications by themselves can worsen cognitive difficulties. Certain music, more specifically Mozart’s Sonata for 2 pianos (KV448) can reduce interictal epileptiform activity and may be a venue to engineer cognitively beneficial music in the future. 36
Self-management programs such as HOBSCOTCH (Home-Based Self-management and Cognitive Training Changes lives) are aimed to improve cognitive problems through problem-solving therapy and learning memory strategies. The program has been proven to improve quality of life and subjective cognition. 37 The program is currently disseminated throughout the Unites States and can be delivered virtually.
Self-management and the self: Partnering with the community to improve outcomes for people with epilepsy
Epilepsy self-management (ESM) refers to the processes used to control seizures and manage the effects of epilepsy. Current approaches for ESM have generally been derived from models of chronic illness self-management. For over a decade, the US Centers for Disease Control and Prevention (CDC) has supported the Managing Epilepsy Well (MEW) Network with the mission of advancing the science related to ESM by facilitating and implementing research, conducting research in collaboration with community stakeholders, and broadly disseminating the findings. 38,39 A variety of evidence-based MEW Network programs have been developed which empower people with epilepsy to improve their health. Over 15 randomized controlled trials assessing different MEW Network delivery modes demonstrate improvements in quality of life, epilepsy knowledge, ESM competency, self-efficacy, and mood as well as reduced seizure frequency. Epilepsy self-management can be implemented in both clinical and community settings, especially when coordinated with epilepsy-focused service agencies. Resources outside of the clinical office can offer important complementary services to what the medical care system can provide and these additional supports may be especially critical for people with epilepsy who live in underserved areas.
Raising the stakes: Complexities for those with neurodevelopmental disorders
For people with developmental and epileptic encephalopathies (DEE) and their families, the wide range of cognitive, behavioral, functional, social, and medical morbidities are overwhelming. These include severe to profound impairments in essential functions such as mobility, hand use, self-feeding, and, perhaps most importantly, communication. 40 Dysregulation of autonomic functions and sleep are pervasive, disruptive to daily life, and highly distressing. Other troublesome medical and neurologic morbidities include hypotonia, dystonia, scoliosis, cerebral-visual impairment, and precocious puberty. The impact on the individual as well as the family can be profound. Most of these concerns are not well-addressed in a care-setting focused on seizures. Most parents report exhaustion, financial stress, and decreased time and attention for their other healthy children. Care is often seen as poorly coordinated and even fractured, especially for older individuals in the process of transferring care or who have completed the transfer of care to adult settings. The need for competent multidisciplinary care with excellent communication among providers (including the family) is paramount for DEE-affected individuals yet is rarely available. Community resources such as DEE-P (Developmental Epileptic Encephalopathy-Project) Connections 41 provide critical, otherwise unavailable information for affected families and care providers. In many ways, it is the families who are leading the way in educating the providers.
Presidential Symposium
Seizure Semiology: The Jacksonian March to the Present
*R. Edward Hogan, MD, Neda Bernasconi, MD, PhD, Hal Blumenfeld, MD, PhD, Mark J. Cook, MD, Jacqueline A. French, MD, Terence J. O’Brien, MD, FRACP
*Chair
John Hughlings Jackson was appointed as a physician at the National Hospital for the Paralyzed and Epileptic in London from 1862 to 1906. Using primarily clinical observations, he established many of the concepts of modern neurology. 42 In addition to his descriptions of focal motor seizures, currently known as Jacksonian seizures, he outlined concepts of focal epileptic seizures, excitation and inhibition during seizures, and temporal lobe epilepsy (which he called the “dreamy state”). Given Hughlings Jackson outlined these concepts without modern tools such as EEG and neuroimaging, his work highlights the importance of interpretation of signs and symptoms (semiology) in the diagnosis of epileptic seizures.
The symposium reviewed, from a modern perspective, original Jacksonian concepts of focal and generalized epileptic seizures, excitation and inhibition during epileptic seizures, and localization of temporal lobe seizures. Further lectures highlighted the importance and role of clinical semiology today, including the role of semiology in clinical trials, and modern approaches to exploring semiology and seizures.
Focal versus generalized epilepsy: Are all epilepsies focal?
The concept of the dichotomization of seizures and epilepsies into generalized and partial (or focal) seizures, while dating back to Hughlings Jackson’s time, did not become common usage until developed by the Commissions on Classification of the International League Against Epilepsy. However, in clinical practice, this dichotomy between generalized and focal epilepsies is not always so clear. Patients with otherwise typical Idiopathic Generalized Epilepsy (IGE) do not uncommonly manifest “focal” clinical and EEG features. 43 In-vivo electrophysiology studies in rodent models of IGE, and human functional MRI studies, have both demonstrated that apparently, generalized absence seizures can originate in a focal region of the cortex before spreading to engage widespread bilateral thalamocortical structures. 44 However, the dichotomy between “Focal” and “Generalized” epilepsy does have clinical value, being important in selecting treatment options, prognostication, and genetic counseling. Differentiation between them is usually possible with expert epileptological assessment. There is also building evidence for an etiological dichotomy between Generalized and Focal epilepsy from large-scale genomics studies. 45
Excitation and inhibition during temporal lobe epileptic seizures
Temporal lobe seizures disrupt normal brain function through abnormal excitatory activity in limbic circuits. This activity also produces inhibition of regions outside the limbic system. The network inhibition hypothesis proposes that inhibition of subcortical arousal systems including the thalamus leads to depressed cortical function and impaired consciousness in temporal lobe seizures. 46 The network inhibition hypothesis is supported by human cerebral blood flow and intracranial EEG studies as well as by experimental animal models. Neurostimulation in epilepsy animal models and in patients with chronic disorders of consciousness targeting the intralaminar central lateral (CL) nucleus of the thalamus demonstrates the feasibility of restoring arousal. 47 Based on this, the START (Stimulation of the Thalamus for Arousal Restoral in Temporal lobe epilepsy) clinical trial is aimed at responsive stimulation of thalamic CL in the hope of improving consciousness during and after temporal lobe seizures that cannot be stopped by other treatment approaches.
Neuroimaging of temporal lobe epilepsy
Hughlings Jackson’s ideas have helped direct the use of MRI as a research and clinical tool. His observations most relevant to neuroimaging of temporal lobe epilepsy include linking symptoms of the “dreamy state” to lesions of the mesiotemporal lobe structures. He also conceptualized epilepsy as a model to understand brain organization. Today, structural MRI reliably detects mesiotemporal sclerosis and pattern learning characterizes its heterogeneity across individuals. Combining whole-brain features of grey- and white-matter pathology with unsupervised topic modeling provides a description of individual variability that refines predictors of clinical outcomes, including drug response and postsurgical seizure outcome. 48 Imaging markers of connectivity have identified large-scale anomalies of brain organization that are at the basis of cognitive impairment across multiple domains. 49
Semiology in clinical trials
Hughlings Jackson was a proficient diagnostician before EEG and neuroimaging were in existence. 50 Epilepsy is among the few diseases that are diagnosed primarily on semiology, with assistance from diagnostic testing. Thus, understanding semiology is critical in classification and appropriate subject selection for clinical trials. We can only study the impact of interventions on specific seizure types and syndromes, if appropriately classified subjects are enrolled. Remote adjudication of trial subjects by the Epilepsy Study Consortium has placed a spotlight on substantial variability in phenotyping from person to person, from center to center, and from country to country, which potentially can lead to enrolling subjects inappropriately. Misclassification of tonic–clonic seizures is of particular concern since they are a seizure type associated with specific harms, including SUDEP. While testing remains important, semiologic assessment remains critical in the determination of eligibility for clinical trials.
Modern approaches to exploring semiology and seizures
Seizure semiology is an essential component of clinical assessment. There is a structure to the development of the features of a seizure that provide a way of understanding how the origin of the event relates to the clinical manifestations, and this ultimately provides a means of seizure onset localization and helps guide management. 51 Hughlings Jackson correlated his close clinical observations of seizure semiology with detailed pathological studies, recognizing the cortical origin of seizures, and that the clinical manifestations were determined by the site of seizure origin and extent of seizure spread. These remarkable insights provided us with the tools we still use today in clinical practice. Advances in intracranial neurophysiology—particularly SEEG—and structural imaging have allowed the relationship between clinical manifestations, seizure onset, and spread to be further clarified, and quantitative methods of analysis such as motor activity through automated video analysis may further improve assessment. 52
Advanced Practice Providers Symposium
Medication Dilemmas: Practical Approaches to Pharmacological Management of Epilepsy
*Kelly R. Conner, PhD, MMS, PA-C, Nancy Auer, APRN, FNP-BC, Danielle A. Becker, MD, MS, Shivani Bhatnagar, DNP, RN, CPNP, Michael A. Gelfand, MD, PhD, Elizabeth H. Michael, MMS, CPNP, CSN, Michelle W. Welborn, PharmD
*Chair
The commitment to care for a loved one with drug-resistant epilepsy often results in substantial emotional, psychosocial, and financial burdens on the entire family unit. Early discussion with caregivers regarding the potential uphill battle in obtaining acceptable seizure control; comorbidities, including SUDEP; and the level of care needed to support the patient is encouraged. Timely referral to family counseling; genetic counseling when appropriate; state-based Early Intervention Programs 53 ; physical, occupational, and speech therapy; and skilled nursing agencies when appropriate is important.
Providers who exhaust all FDA-approved ASMs and seek better seizure control may consider accessing investigational drugs through FDA Expanded Access 54 or Personal Importation programs. State-based Medicaid waivers provide Medicaid, in-home skilled nursing, and nonskilled care for medically fragile and intellectually disabled children and adults, regardless of parental income or primary private health insurance coverage. Letters of Medical Necessity often make or break access to medications and services.
Choosing the first anti-seizure medication
Choosing the first anti-seizure medication for a patient with newly diagnosed epilepsy can be daunting considering there are currently over 30 available medications. The ideal anti-seizure medication would maximize seizure freedom without unwanted side effects.
Full seizure control can be achieved in about 50% of patients and about 30% will be refractory to medication treatment. 55 The success rate depends on several factors including the type of epilepsy and family history.
Factors influencing the appropriate choice include epilepsy classification, patient age, comorbid conditions, current medications, potential side effects, time to titration, dosing regimen, and cost.
It is important to use a targeted approach and choose the medication best matched for the epilepsy type. If the focus is unknown, use a broad-spectrum ASM. If the patient has comorbid conditions (such as headache), consider a medication that will treat underlying epilepsy as well as the comorbid condition. 56
Polypharmacy, is less more?
Drug–drug interactions are common and thus one should be aware of pharmacokinetic and pharmacodynamic interactions and choose drug selection accordingly. Utilization of various mechanisms of action is KEY to avoid side effects from pharmacokinetic interactions. 57 If a new medication is started and is more efficacious, evaluate and possibly reduce previous treatment to improve tolerance and retention. It was also discussed that the use of lower doses and slower titrations in the elderly may improve tolerance and reduce side effects.
Consider nonpharmacologic methods: surgery, neuromodulation, and diet therapy. Neuromodulation does not produce the same side effects as medication and in fact has shown improvements in cognition, memory, and mood. The use of neuromodulation can help reduce polypharmacy. The data from the RNS allows providers to identify the efficacy of new medication early to determine clinical benefit of titration and possibly guide the reduction of other anti-seizure medication. 58
Thus, it is important to consider reducing medications after surgery or neuromodulation devices have been placed. By reducing medication, one can reduce side effects and ultimately improve quality of life.
Drug interactions and medications that lower seizure threshold
Interactions between ASMs and other medications (non-ASMs) can be significant. Interactions may be pharmacodynamic, however, pharmacokinetic interactions may be more meaningful. Enzyme-inducing ASMs may decrease non-ASMs efficacy. Conversely, enzyme inhibition can increase non-ASM side effects. Likewise, non-ASMs can cause either decreased ASM efficacy or increased concentration. Management of interactions depends on the duration of use and the availability of ASM and/or non-ASM alternatives. 59
Non-ASMs can also “lower the seizure threshold”; either inducing a seizure in a patient without epilepsy or worsening seizures in patients with epilepsy. When considering medication combinations, questions include: Necessity? How urgent is the need for treatment? How strong is the seizure-provoking effect? Are alternatives available? Avoidance of the non-ASM, short-term “bridge” or titration of ASM may be appropriate. Medication classes of common concern include antibiotics, antidepressants, and antipsychotics. Of note, most common antidepressants, with limited exceptions, are considered safe at therapeutic doses. 60
Seizure action plans and rescue medications across the lifespan
Most seizures occur outside of a hospital with few persons with epilepsy having seizure action plans (SAP). An SAP is an aide for seizure care.
Seizure action plans are effective in providing confidence to parents regarding their child’s epilepsy diagnosis, increasing the return rate of neurology appointments, and are more useful for people with lower seizure frequency. They are not effective in decreasing ED visits or health care utilization. It may not be the SAP itself, but the education and training of the individuals who institute SAPs. States are requiring education and training of school staff on seizure care. Currently, how improved knowledge affects health care utilization is being studied.
Several rescue medications are commonly incorporated into SAPs: Intranasal midazolam and diazepam, and rectal diazepam. They are safe, effective, and easily administered.
In conclusion, SAPs allow persons with epilepsy to participate in societal normal activities with safe and effective rescue medications. 61,62
Special situations: Surgery/NPO, illness, missed dose and so on
Shivani Bhatnagar, DNP, RN, CPNP, discussed how to formulate a plan and choose appropriate alternatives for anti-seizure medications (ASMs) in special situations. She summarized ASMs can be safely given by mouth with a small amount of water when a patient is NPO, for extended NPO status, an alternative ASM plan should be in place. For a missed medication dose, if it has been less than half the time between doses (i.e., for once-a-day dosing that is 12 or more hours before the next dose) the patient can be advised to safely take the dose. If it has been longer than half the time, the patient should be advised to wait to take the next scheduled dose. 63 She highlighted 2 key points regarding the ketogenic diet, in the setting of illness: (1) preventing, monitoring, and treating for hyperketosis and (2) maintaining hydration, take priority over maintaining ketosis. 64
Epilepsy Therapies Symposium
New Approaches to Drug-Resistant Epilepsy
*Dean Naritoku, MD, *Alica M. Goldman, MD, PhD, Allyson Alexander, MD, PhD, Danielle Andrade, MD, MSc, FRCPC, Ingmar Blümcke, MD, David Burdette, MD, Stephan Schuele, MD, MPH, FAES
*Co-Chairs
An estimated 400 000 of the 2 million individuals with focal epilepsy in the United States are medically refractory 65 with resultant disabling seizures, increased risks of premature death, injuries, psychosocial dysfunction, and reduced quality of life for the patients and their caregivers. 66 The effect of a drug-resistant epilepsy (DRE) on personal and professional achievement or employment, the need for long-term treatment, and the often recurrent hospitalizations contribute to a considerable individual and societal economic burden. 67 Remediation of DRE is complex. However, novel molecular diagnostic approaches in neuropathology are aiding in precision diagnostics, discoveries in epilepsy genetics are shedding light not only on the DRE causality but also on comorbidities and associated adverse outcomes, and new anti-seizure medications and surgical approaches offer hope to many patients. The 2022 Epilepsy Therapy Symposium aimed to address some of these advances as relevant to clinical practice.
Impact of genetics on surgical management of epilepsy—Advancements in neuropathology
Considerable advances in understanding the genetic causes of cortical malformations and low-grade epilepsy-associated brain tumors (LEAT) prompted the adaptation of our current disease classification systems integrating genotype–phenotype associations. 68 Such genotype–phenotype association is established for somatic (postzygotic) mutations related to the mTOR pathway in Focal Cortical Dysplasia ILAE Type II located predominantly in the frontal lobe. 69 Another common association is that of altered MAP-kinase signaling in LEAT predominantly located in the temporal lobe, 69 or brain somatic mutations in the galactose transporter gene SLC35A2 in mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE). 69 -71 The latter association also helped to define MOGHE as a new disease entity 68 and postsurgical seizure freedom rates increased from 33% in the initial report 71 to 64% in a most recent study. 70 This knowledge will finally also help to move toward precision medicine, that is, D-galactose supplementation in patients with MOGHE.
New anti-seizure medications: Update for 2022
The years 2019 to 2022 have seen the approval of 3 anti-seizure medications with unique, putative mechanisms of action driving unique spectra of efficacy. Cenobamate was approved in 2019 for the treatment of focal (partial) seizures in adults. Cenobamate is a positive allosteric modulator of GABAA receptors and inhibits the persistent component of the sodium current. It is quite effective but requires a slow titration to minimize the risk of drug reaction with eosinophilia and systemic symptoms (DRESS). 72,73 Fenfluramine was approved for the treatment of seizures associated with Dravet Syndrome in 2020 and for Lennox–Gastaut syndrome in 2022. It reduces serotonin uptake and increases serotonin release thereby producing seizure reduction, appetite reduction, and rarely potential cardiac valvulopathy. 74,75 Ganaxolone was approved for treating seizures associated with a rare genetic disorder, CDKL5 deficiency disorder. It activates synaptic and extrasynaptic GABAA receptors producing specific efficacy in this developmental and epileptic encephalopathy. 76
New approaches for treatment of multifocal epilepsy
Multifocal epilepsy includes a wide spectrum of disorders that can be defined either by semiology, neurophysiology, or etiology as having potentially independent or widespread foci of epileptogenicity. Based on etiology, multifocal epilepsies include patients who failed surgery are found to have a germline or somatic mutation, lack an MRI abnormality, or demonstrate multifocal lesions on imaging such as tuberous sclerosis, multiple cerebral cavernoma syndrome or patients with bilateral nodular heterotopia, polymicrogyria or hemi-megalencephaly. Select patients with consistent focal features in their presurgical workup, in particular a localizing semiology and regional seizure onset pattern, can be candidates for an invasive evaluation using SEEG. 77 For patients with multifocal epileptic encephalopathies, neuromodulation is a consideration targeting the centromedian nucleus of the thalamus in addition to more traditional stimulation targets.
Minimally invasive epilepsy surgery techniques
Clinical studies have repeatedly shown that patients with drug-resistant epilepsy are unlikely to become seizure free with traditional anti-seizure medications alone and they ought to be considered for epilepsy surgery. Modern techniques in minimally invasive epilepsy surgery offer options for many patients 78 and fall into 4 main categories: diagnostic, ablative, disconnective, and neuromodulatory. Within the diagnostic category: SEEG has become a widely accepted technique for phase II monitoring. Minimally invasive forms of ablation for epilepsy include laser interstitial thermal therapy (LITT) and radiofrequency (RF) ablation. Focused ultrasound is a newer technique for ablation of brain tissue that may prove efficacious for the treatment of epileptogenic lesions in the future. Minimally invasive techniques for disconnection of broad epileptogenic networks include the use of endoscopy or LITT to accomplish corpus callosotomy and hemispherectomy. Finally, neuromodulation for epilepsy includes vagus nerve stimulation, deep brain stimulation, and responsive neurostimulation. 79 Advantages of minimally invasive approaches include smaller incisions, decreased surgical complications, reduced hospital stay, increased palatability for patients and families, and expanded indications for patients with generalized epilepsies.
Genetic approaches to epilepsy comorbidities
Understanding the genetic etiology of epilepsy syndromes is important not only for precision diagnostics but also to guide targeted therapies. Comorbidities are an important aspect of many epilepsies. They may occur independently of epilepsy (i.e., cortical visual impairment in CDKL5-related epilepsy), consequently to epilepsy (epileptic encephalopathy at the onset of treatment-resistant seizures), or be treatment-related (sedation and cognitive slowing in the context of polypharmacy). Design of targeted therapies ought to consider these aspects as the optimal outcome will remediate both epilepsy and related comorbidities. Examples of therapies where multifaceted benefits have been observed are (1) fenfluramine that aids in seizure control but may also reduce mortality 80 and lead to an improvement in executive function in patients with DS, 81 (2) everolimus, an mTOR inhibitor that helps control growth progression of subependymal giant cell astrocytomas (SEGAs), improves seizure control, and possibly symptoms of autism in patients with tuberous sclerosis complex. 82,83 Gene allele-specific antisense oligonucleotide therapies have been emerging as a new line of promising treatment strategies.
Treatment-resistant epilepsies affect an estimated 30% of patients and their treatment represents an ongoing important challenge. However, investment in basic and translational research has driven annual progress in genetics, precision molecular diagnostics, minimally invasive probing of epileptic networks, and novel surgical therapies. Therapeutic focus has expanded beyond seizure suppression toward striving to minimize drug-related adverse effects and to combine epilepsy control with positive effects on comorbidities and adverse outcomes, such as SUDEP.
Best Practices in Clinical Epilepsy Symposium
Access to Care for the Underserved Managing Epilepsy
*Shanna Guilfoyle, PhD, *Madona Plueger MSN, ACNS-BC, CNRN, Lisa Clifford, PhD, Jeannine Conway, PharmD, Jasmine Kwasa, PhD, Sarita Maturu, DO, Christopher Ryan, MSW, LICSW, Lindsay M. Schommer, MSN, APRN, ANP-BC, Naymee Velez-Ruiz, MD, Claire Waller, Jill Waller
*Co-Chairs
Madona Plueger APRN, CNRN, FAES introduced the symposium and the content experts presenting topics that address both clinical and research integration of care for underserved and vulnerable populations managing epilepsy from the interdisciplinary lens. Our symposium sets out each year to provide key implications for practice not only across the continuum of care of the person with epilepsy but also across the landscape of care that is available. The goal of this symposium is to capture opportunities applicable from the smallest to largest institutions, being able to apply principles to practice.
Claire and Jill Waller; patient advocates, opened our symposia by sharing Claire’s story of the initial diagnosis and provided a glimpse to us, of the journey of a person and family living with epilepsy. The richness of the candid reality of epilepsy as a disease, coupled with the triumphs and tribulations along the way, was a perfect way to meld the lectures together.
Lisa Clifford, PhD, presented “Defining Social Determinants of Care in Vulnerable Individuals with Epilepsy.” The presentation addressed the importance of understanding the social determinants of health conceptual framework and health inequities in epilepsy. This presentation was rich with key implications of care and data-driven support, validating the ongoing concerns of social determinants of health in the field of epilepsy. 84 Dr Clifford challenged us all to work on continuing to close the treatment gap. This will require a multisystem, integrative approach that addresses inequities at all levels.
Lindsay Schommer APRN and Christopher Ryan MSW, LICSW took on the task of sharing information on barriers to access to care. Ms. Schommer addressed accessibility to care in her presentation “Access to Epilepsy Care in Rural Communities: Challenges and Opportunities.” Ms. Schommer addressed the barriers to access for those living in communities and some thoughts on how telemedicine and other venues of access to care are approached. 85 This presentation reminded all of us about some things that most take for granted. Things like distance, transportation availability, and accessible internet. Health literacy concerns are higher in rural communities than in other areas. Opportunities on other networks, such as support groups, church support, and resources for self-management, were shared. The Epilepsy Community has formed several established self-management programs. Ms. Schommer spent a few minutes and shared the definitions and brief overviews of Hopscotch, Uplift, Paces, Mindset, Time, Pause, and Smart. These programs are available throughout the country; some in communities with limited resources. Mr. Ryan’s presentation focused on recognition and awareness of the importance of psychosocial screening design and some of the barriers to care in their use. Practical examples of how these screeners can be used across the continuum of care were shared. Mr. Ryan shared that there may be opportunities to use semi-structured psychosocial screens to complement empirically validated instruments that are used clinically. The semi-structured approach offers an opportunity for assessment and direction as to when to refer to psychosocial providers for proactive screening. He left the audience with the understanding that high acuity new onset population benefit from automatic referrals.
Jasmine Kwasa, PhD, addressed the attendees from the basic science lens of EEG lead application and assessment of hair preparation of black individuals, as a backdrop to a poignant discussion on the focus on the importance of cultural competency. Dr Kwasa addressed how the cultural competency approach improves inclusion in basic and clinical research through approaches in hiring inclusive medical staff. Dr Kwasa provided an example by addressing research being completed on traditional EEG systems on afro-textured hair-prep and tech solutions. 86 Discussion of a current process and protocol for hair preparation provided attendees with evidence on avoiding patient discomfort, increasing medical trust in marginalized communities, and therefore increasing data fidelity.
Jeannine Conway, PharmD, presented “How to Get Bang for Your Buck with Anti-Seizure Medications.” This session provided practical points for providers that are faced with the task of getting the right medications for seizure control, for those uninsured. Basic Information was shared, along with strategies to optimize anti-seizure medication costs for persons with epilepsy with payer challenges. Often providers of care struggle to be able to articulate availability resources. Different countries and different parts of the country set medication prices. The lecture also included the whys that are behind the scenes with drug coverage. This included discussion about the regulatory bodies and venues of payment, such as mail order, pharmacies, drug coupons, and so on.
Claire and Jill Waller returned to the podium. The story continued as Claire shared the surgical workup, surgery intervention, and now celebration of seizure freedom for a duration of time. A poignant message resonated from Ms. Jill Waller, as being the mother of a vibrant young lady who advocates strongly for epilepsy awareness. A person may be seizure-free, but the person continues to have epilepsy, which affects the entire family and surrounding community. The importance of advocating with that message rang through the room as the ebb and flow of motions of a person and family with epilepsy were shared.
Sarita Maturu, DO, and Naymee Velez Ruiz, MD, copresented on the models of care for women with epilepsy in their presentation “Insights and Suggestions for Adopting Models of Care for Pregnant Women with Epilepsy.”
Dr Maturu presented resource-rich and resource-spare opportunities in opening up a pregnancy clinic for women with epilepsy and shared insights on how to create and sustain such a clinic. The importance of establishing relationships early on, benefits immensely, assisting in sustainability with assuring that key stakeholders are aware of services and support to women with Epilepsy that become pregnant. Dr Velez Ruiz shared data supporting the models of care. 87 Participants were able to take key implications of practice away with the understanding that data can drive change and the viability and sustainability for such clinics.
Co-Chairperson Shanna Guilfoyle, PhD, invited speakers back to the stage and recognized our advocates, as well as the speakers overall. Audience participants were able to address questions and discussions continued after the completion of this symposium.
Epilepsy Surgery Symposium
Epilepsy Surgery Controversies: A Case-Based Discussion
*Guy M. McKhann II, MD, *Jorge Gonzalez-Martinez, MD, PhD
*Co-Chairs
The 2022 Epilepsy Surgery Symposium focused on current state-of-the-art methods and procedures related to epilepsy surgery, with particular attention to the main controversial topics in the field. Controversies discussed included (1) open resection versus laser ablation for mesial temporal lobe epilepsy; (2) surgical approaches to lesional neocortical temporal lobe epilepsy; (3) responsive neurostimulation (RNS) versus deep brain stimulation (DBS) for primary generalized epilepsy; and (4) SEEG versus subdural grid monitoring and mapping for dominant perisylvian epilepsy. After case presentations introducing each topic, speakers presented the different surgical approaches, discussing indications, advantages, and limitations for the respective approaches and clinical scenarios. After the presentations for each case, a controversy-based session took place, motivating intense discussion and participation from the audience.
Following participation in Epilepsy Surgery Symposium, participants are able to discuss the different aspects of epilepsy surgery practice, including indications, techniques, and expected results from different approaches to specific clinical scenarios. For the clinical topics that were presented, participants recognize the challenges and controversies related to surgical interventions, the options available, and the advantages and disadvantages of each intervention. Participants further recognize the value of epilepsy neurology and neurosurgery’s close clinical and surgical collaboration, promoting positive patient outcomes and minimizing adverse consequences.
The program started with a short introduction of Speakers and learning objectives followed by Dr Arka Mallela’s presentation of an amygdala-centered mesial temporal lobe epilepsy (MTLE). Subsequently, Dr Guy McKhann exposed the differences in indications and techniques related to standard versus selective resections for MTLE, emphasizing the importance of individualization of care. Dr Chen Wu then discussed applying laser ablation (LITT) therapy to the presented case and to MTLE in general. The presentations were followed by a debate among speakers and the audience, focusing on the advantages and disadvantages of each approach.
The second case was presented by Dr Garrett Banks, an example of lesional neocortical temporal lobe epilepsy. The case presentation was followed by Dr Stephen Ojemann’s talk, discussing the indications and techniques of neocortical temporal resections guided by intraoperative monitoring with electrocorticography (ECOG). Subsequently, Dr Brett Youngerman discussed the advantages and challenges of utilizing extraoperative invasive monitoring in defining the epileptogenic zone in lesional neocortical temporal epilepsies.
The third case, an example of primary generalized epilepsy, was presented by Dr Hussam Shaker. This presentation was followed by a discussion focusing on the utilization of closed-loop RNS Stimulation by Dr Mark Richardson, in which he summarized methods and techniques related to RNS thalamic stimulation in generalized epilepsy, particularly focusing on his experience with centromedian thalamic closed-loop stimulation. Subsequently, Dr Arthur Cukiert, presented his experience in the utilization of deep brain stimulation (DBS) open-loop stimulation therapy in patients with generalized epilepsy.
The final topic of the symposium focused on the surgical management of nonlesional focal epilepsies and the advantages and disadvantages related to the different methods of invasive monitoring. Dr Jessica Fessler began the discussion, presenting a case of nonlesional dominant perisylvian epilepsy, with rich semiological features of auditory auras followed by generalized tonic–clonic seizures. Dr Jorge Gonzalez-Martinez next discussed the indications and results of utilizing the SEEG methodology in nonlesional epilepsies, emphasizing the importance of SEEG-based spatial-temporal dynamics and 3-dimensional mapping of the epileptogenic zone. Dr Yemi Damisah then discussed the indications, advantages, and disadvantages of using subdural grids in mapping the epileptogenic zone and performing the functional mapping.
The Epilepsy Surgery Symposium was finalized with a session of questions and answers related to the presentations, with an engaging dialogue among the speakers and audience.
Annual Course
Epilepsy in the Era of Personalized Medicine
*Kelly Knupp, MD, MSCS, *Heather R. McKee, MD, Allyson L. Alexander, MD, PhD, Jacquelyn L. Bainbridge, PharmD, FCCP, MSCS, Sallie A. Baxendale, PhD, Robyn M. Busch, PhD, Cornelia Drees, MD, Taneeta Mindy Ganguly, MD, Tracy A. Glauser, MD, Ann Hyslop, MD, Jong Woo Lee, MD, PhD, Kimford J. Meador, MD, M. Scott Perry, MD, Shilpa B. Reddy, MD, Jessica W. Templer, MD, Sophia M. Varadkar, MRCPI, MSc, PhD, Zhong Irene Wang, PhD
*Co-Chairs
Personalized medicine is a refined and optimal treatment approach. The Annual Course addressed the personalized management of epilepsy from unique perspectives and populations. This included topics on genetic testing, potential gene modulatory therapies, pregnancy management, and epilepsy associated with oncologic processes. Pharmaceutical management, personalized prediction of surgical outcomes, bias in surgery, and surgical approaches were also addressed in the comprehensive course. A thread throughout the course was a patient’s mother who addressed her personal story regarding genetic testing, medication experience, and surgery with her child’s genetic epilepsy syndrome.
The first session focused on genetics, including ordering genetic testing, understanding results, and gene modulatory therapies. A summary of this section is detailed in a separate summary publication.
The second session addressed personalized pharmaceutical management, including detailed and careful approaches in pregnancy, oncology patients, and pharmacogenomics. A debate about polypharmacy closed the session.
Dr Kimford Meador opened session 2 with a lecture on personalized pharmaceutical management for patients with epilepsy during pregnancy which elaborated on the following main points. Women with epilepsy (WWE) should receive informed consent outlining risks before conception, preferably when anti-seizure medication (ASM) is first prescribed and repeated at least yearly. Although increased risks exist, most children born to WWE are normal. Risks for many ASMs are uncertain, but valproate is a poor first-choice ASM for most WWE of childbearing potential. Lamotrigine and levetiracetam have safer profiles. Women with epilepsy of childbearing potential should be on folate. Clearance changes in pregnancy occur with many ASM requiring increased doses to maintain pre-pregnancy levels and then adjusted back in postpartum. Anti-seizure medication dosing in pregnancy needs to balance seizure control and risk to the fetus. If dose adjustments are done to correct clearance changes, pregnant WWE is no more likely to have increased seizures during pregnancy than nonpregnant WWE. Mood and anxiety disorders should be assessed during pregnancy and postpartum and treated if present. Breastfeeding on ASMs appears safe. 88,89
Following this, pharmacogenomics, and its impact in the field of epilepsy was presented by Dr Tracy Glauser. Genetic variability can impact ASM pharmacokinetics and/or pharmacodynamics resulting in unexpected changes in efficacy, dose-dependent side effects, or idiosyncratic reactions. 90 For a few ASM, there are 4 well-characterized anti-seizure medication pharmacogenetic relationships (CYP2C9, CYP2C19, HLA-B*1502, and HLA-A*3101). 91,92 The most cited one is between the HLA-B*1502 allele and carbamazepine, oxcarbazepine, and phenytoin/fosphenytoin with increased risk in patients of Asian descent (other than Japanese or Korean descent) of toxic epidermal necrolysis and Stevens–Johnson syndrome. 91,92
Important current challenges to implementing routine pharmacogenetic testing in outpatient epilepsy clinics include (i) variability in ASM drug response is more often related to drug–drug interactions rather than genetics, (ii) few ASM have a genetic component in their pharmacokinetic pathways so prescribers have multiple ASM options not affected by genetics, and (iii) in routine clinical practice, risk of idiosyncratic reactions is less important in driving prescribing behavior than ASM efficacy and dose-dependent toxicities.
In the future, a holistic computational trajectory approach integrating genetic data, drug–drug interactions, demographic data, and environmental data could be impactful in reducing the variability in ASM response and optimizing our patients’ seizure control and quality of life. 93
Dr Jessica Templer provided her expertise on personalized pharmaceutical management for multidisciplinary care in neuro-oncology and epilepsy. Managing seizures in patients with brain tumors presents unique challenges that the treating provider should consider when making treatment decisions. It is essential to recognize the impact of seizures as well as the side effects of anti-seizure medications for patients with tumor-related epilepsy.
The frequency of seizures is most significantly related to tumor type with low-grade tumors associated with a higher risk of seizures and high-grade tumors often carrying a relatively lower risk of seizures. While anti-seizure medications may potentially be associated with side effects including poor cognition or metabolic dysfunction, the impact of seizures as a reminder of a patient’s brain tumor cannot be understated in this population. It is essential to review the patient’s seizure frequency, individual tolerability of their anti-seizure medication regimen, and the patient’s unique goals at each clinic visit to ensure the optimal treatment for each individual patient. 94,95
To close the session, Doctors Lee and Bainbridge discussed opposing sides of whether polypharmacy is beneficial or not in pharmaceutical management.
Dr Jong Woo Lee presented the point of view that “best medicine is less medicine.” Polypharmacy is typically defined as 5 or more medications, with at least one inappropriate medication. In patients with epilepsy, 37% use at least 2 ASMs. Polypharmacy increases with age and has also increased over the past 30 years. There is greater variation in prescribing related to prescriber factors than patient factors. 96
Polypharmacy magnifies the hazards associated with monotherapy. The most significant hurdle of polypharmacy is medication side effects. Drug–drug interactions may cause unfavorable pharmacokinetic and pharmacodynamic interactions. Although there are examples of synergistic effects of ASM, there are numerous potential antagonistic effects. Polypharmacy increases noncompliance; using 2 ASMs increases odds of nonadherence by 30%; using 3 or more medications increases odds 2- to 3-fold. 97 Polypharmacy increases the risk of medication errors, particularly in the elderly. Lastly, polypharmacy is inevitably associated with an increase in medication costs.
When polytherapy is required for optimal seizure management, extreme care should be exercised to minimize these risks.
In response to this, Dr Bainbridge presented the opposing point of view that polypharmacy can be beneficial when treating patients with epilepsy.
For many patients, epilepsy is complicated. In 60% to 70% of patients with epilepsy, treatment with a single ASM, “monotherapy,” provides adequate seizure control. For 30% to 40% of patients, monotherapy isn’t effective, denoted drug-resistant epilepsy (DRE). Each patient’s epilepsy is unique, and DRE requires a patient-centered “personalized medicine” approach. Consider each patient’s adverse effects, epilepsy etiology, comorbid conditions, drug costs, and adherence.
Combining multiple, mechanistically unique ASM, “rational polytherapy,” is particularly useful for treating DRE. Some ASM combinations are synergistic, valproate, and lamotrigine most famously so. Leveraging pharmacokinetic and pharmacodynamic interactions can improve efficacy, potentially allowing the use of lower doses. Conversely, avoiding combinations with identical mechanisms can make adverse effects less likely. Indeed, treatment failure due to adverse effects following the addition of a second ASM may not differ from monotherapy alone.
Rational polytherapy by individualizing care is our best chance to help the 30% to 40% of patients with DRE. 98,99
The third session focused on personalized preparation for surgery and included an enlightening debate on surgical outcomes, as well as lectures on predicting cognitive and mood outcomes, and addressed bias in epilepsy surgery.
Dr Cornelia Drees presented the stance of nonseizure outcomes being more important regarding epilepsy surgery. Nonseizure outcomes are neglected measures of success—or failure—after epilepsy surgery. Patients and physicians focus on seizure freedom while overlooking other major determinants of quality of life (QOL). This view disregards the greater impact that psychosocial distress, loneliness, and struggles with adjustment and stigma have on overall QOL compared to seizure frequency. 100 Yet, “seizure freedom” represents the hope for independence, opportunity, health, and happiness. Reaching these goals is easier when seizures are controlled, though they should be pursued despite seizures. And, despite seizure freedom, patients can continue to be challenged by the “burden of normality” and changes in family dynamics. 101 These potential difficulties need additional support for patients and families in both the pre- and postsurgical phase. A multidisciplinary team that includes a psychologist, psychiatrist, and social worker could address patients’ life goals and hardships parallel to attaining seizure freedom.
On the contrary, Dr Sophia Varadkar presented the view that seizure outcome is a fundamentally important factor with regard to epilepsy surgery. Resective epilepsy surgery is the most clinically effective treatment for children, young people, and adults with drug-resistant focal epilepsy. The primary aim is to stop seizures. International League Against Epilepsy (ILAE) expert consensus recommendations are to offer an evaluation to every suitable patient with drug-resistant epilepsy. Early surgery improves outcomes. Patients are carefully selected after multidisciplinary assessment. Shared decision-making with the patient (and family) must include careful discussion of hoped-for-benefits and possible risks, individualized for that patient. Discussion includes likelihood of seizure freedom or meaningful seizure reduction and possibility of seizure return. Surgical mortality is rare, and morbidity is surgery and patient-specific, carefully considering motor function, language, memory, and vision. Seizure freedom may allow reduction of drug burden, all-cause mortality, SUDEP, injuries, and health care resource utilization. It may allow the person to drive and increase life choices and quality of life. In temporal lobectomy, cessation of antiepileptic medication is the strongest predictor of IQ increase. Other focal surgeries may offer modest improvements in IQ. 102
Dr Robyn Busch, PhD, joined us to delve into predicting individual cognitive and mood outcomes following epilepsy surgery. Temporal lobe resection is an effective treatment option for pharmacoresistant temporal lobe epilepsy (TLE) but is often associated with declines in cognition and mood that can negatively impact patient functioning and quality of life. A recent series of multicenter studies developed and validated multivariable prediction models for older adolescents and adults with TLE being evaluated for epilepsy surgery. These models consolidate multiple, often contradictory, risk factors to identify an individual patient’s risk for postoperative declines in language, memory, and mood following temporal lobe resection given their unique demographic and disease characteristics. 103 -105 The models have good to excellent discriminatory ability (concordance statistic range 0.70-0.84) and very good calibration. All models are publicly available in 2 easy-to-use formats—nomograms and online risk calculators—that clinicians can use to estimate the probability of cognitive and mood declines in their patients considering resective surgery for treatment of TLE and to aid preoperative decision-making and patient counseling.
Dr Sallie Baxendale discussed prehabilitation prior to surgery. Postoperative declines in cognitive function following epilepsy surgery can be categorized as cognitive contraindications to surgery, cognitive complications of surgery, and cognitive costs of surgery. A contraindication describes the unacceptable risk of a cognitive deficit developing following surgery, such that surgery is not a viable treatment option for the patient. Cognitive complications of surgery refer to unexpected cognitive deficits which may result from perioperative processes, postoperative events, or an incomplete appreciation of the salience of preoperative information. The cognitive costs of surgery are the expected declines in function associated with the proposed surgery. These costs are different for every patient and can be predicted on an individual basis prior to surgery. Prehabilitation refers to the process whereby cognitive functions are utilized before they are lost to build the compensatory strategies and routines that will be required after surgery to manage the losses anticipated from surgery. 106,107
Bias in epilepsy surgery was presented by Dr Shilpa Reddy. Identifying patient populations overlooked for surgical management of epilepsy is vital in addressing barriers contributing to underutilization of or delay in surgical intervention. Disparities in patient demographics (i.e., race, English proficiency, age), seizure types, epilepsy syndromes, etiology of epilepsy, and psychiatric and cognitive comorbidities have all been cited as biases in epilepsy surgery referrals. The lecture emphasized findings in the article entitled “Underrepresented Populations in Pediatric Epilepsy Surgery.” 108 Addressing disparities in health care, particularly in vulnerable populations, allows us to take one step forward toward narrowing gaps in epilepsy care.
The final session addressed personalized approaches to epilepsy surgery. The session started with a debate detailing about surgery in genetic epilepsy and then addressed tailored imaging strategies, personalized surgical approaches, and the use of devices.
Dr Ann Hyslop led the debate with the perspective of not pursuing surgery in patients with genetic epilepsy. Genetically driven treatments other than anti-seizure medications may reduce seizures and improve associated symptoms including cognition, behavior, and gait in a subset of patients with genetic epilepsies, 109 but phenotypic variability plays a significant role in the efficacy achieved in the individual patient. Thus, there is a need for more data, better phenotyping, consistent functional testing, and further development of targeted therapies. 110 Rather than turn immediately toward surgical interventions when a patient fails 2 appropriately chosen and dosed anti-seizure medications, it is incumbent upon providers to ensure that a genetic evaluation has been done, is complete, and up to date prior to epilepsy surgery in every patient, even in cases of nonacquired focal lesions. By doing so, epileptologists may be able to provide patients with all available options and more accurate prognoses for genetic epilepsies, whether or not surgery is ultimately performed.
Dr Scott Perry presented the opposing perspective for doing surgery on genetic disorders. Epilepsy surgery is an effective, yet underutilized treatment for DRE. Underutilization is particularly true for epilepsies where seizure freedom is not expected, or the etiology remains following surgery (i.e., genetic epilepsies). Surgery for genetic epilepsy can result in significant seizure reduction and improvements in quality of life and development.
Genetic conditions wherein the epileptogenic source is a focal brain malformation (i.e., tuberous sclerosis complex and DEPDC5-related epilepsy) experience seizure-free rates exceeding 50% in many studies. 111,112 Early surgical therapy is also associated with improved developmental trajectories. Conditions without structural epileptogenic sources, such as channelopathies, have been described as poor surgical candidates, yet data is often biased toward the expectation of seizure freedom. 113 Instead, it is imperative to consider the palliative benefits of surgery in this group. Procedures such as corpus callosotomy demonstrate response rates often exceeding those of medications approved for these conditions, in addition to improvements in nonseizure outcomes. 114 Data from the Pediatric Epilepsy Research Consortium’s Surgery Database found two-thirds of patients with genetic etiology experienced >50% seizure reduction following surgery. 115 While nonlesional genetic etiologies were often palliative procedures, 35% had >90% seizure reduction at a mean 11-month follow-up. Therefore, surgery must be considered a viable treatment option for genetic epilepsies in the absence of precision medical treatments with better disease-modifying effects.
Tailored epilepsy imaging strategies for personalized care, focusing on structural MRI, was presented by Dr Irene Wang. The talk centered around 3 important aspects that imaging could help with lesion presence, border, and characteristics. The importance of using dedicated MRI protocol and reviewing guidelines was addressed. The utilization of voxel-based and surface-based MRI postprocessing methods has shown great promise (and in some cases proven essential) for lesion detection and delineation in everyday clinical care. Emerging advanced techniques such as 7 T and MR fingerprinting (MRF) 116 may detect previously unseen lesions in patients with nonlesional 3 T MRI. Lesion characteristics such as FCD subtypes and epileptogenicity may be interrogated by multimodal MRI and MRF. Machine/deep learning studies using multicenter, large data sets, may soon be used for individual detection/prediction on local datasets. 117,118 It is hoped that the integrative use of advanced MRI and postprocessing in everyday care will substantially improve the individualized yield of structural MRI.
Dr Allyson Alexander presented personalized surgical approaches and new tools at our disposal. Epilepsy surgery is an excellent example of personalized medicine. Presurgical workup includes video-EEG, MRI, MEG, PET, SPECT, and/or phase II evaluation to localize the seizure focus for each individual patient. If the seizure focus is resectable, traditional open surgery or laser interstitial thermal therapy may be performed to remove the epileptogenic zone. Patients with generalized or multifocal epilepsy have traditionally had few surgical options. Currently, many such patients are eligible for neuromodulation (summarized in Table 2). Vagus nerve stimulation (VNS) is a simple extracranial outpatient procedure with minimal risks; however, there are few data available to help predict which patients will respond to this therapy. Intracranial forms of neuromodulation, including deep brain stimulation (DBS) and responsive neurostimulation (RNS) offer more customizable forms of therapy via targeting of neuromodulatory signals to the patient’s seizure disorder. Current research is underway to develop biomarkers, such as an individualized brain connectome, that will allow the prediction of an optimal form of neuromodulation for each patient. 119 -122
Comparison of Currently Available Forms of Neuromodulation.
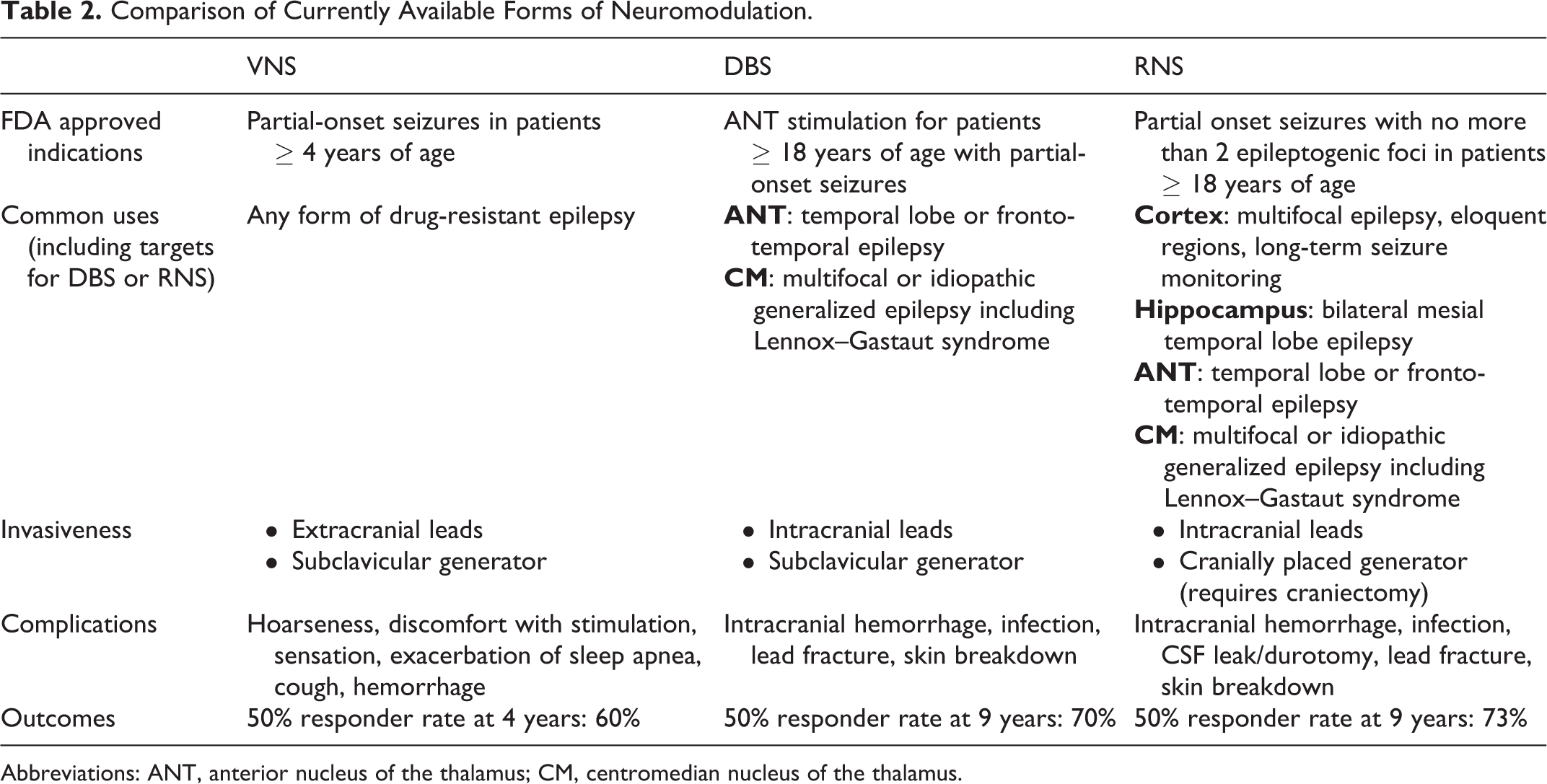
Abbreviations: ANT, anterior nucleus of the thalamus; CM, centromedian nucleus of the thalamus.
Dr Ganguly joined with a discussion on the potential of standard-of-care techniques in personalizing therapeutic device selection for those with drug-refractory epilepsy. For example, functional connectivity on MEG and network synchronizability on SEEG are calculated measures that have demonstrable differences between responders and nonresponders to RNS. While biomarkers like these are not yet ready for widespread clinical use, they show promise—some of these predictive functional neuroimaging techniques have been used to prospectively predict response to VNS at independent enrollment sites. Dr Ganguly also outlined the role of long-term diagnostic devices and wearables in optimizing quality of life for those with drug-resistant epilepsy. Long-term subscalp and intracranial EEG have been used to successfully forecast periods of high seizure risk. These prediction models are generalizable, meaning that a classifier trained on a group of patient data is able to successfully predict seizures in a different, yet unseen group of individuals. Aiming to define “pro-ictal states” of high seizure probability allows for the integration of multimodal data including accelerometry, electrodermal activity, and surface EMG which have acceptable efficacy in commercially available wearables. 123,124
The 2022 American Epilepsy Society Annual Course on Epilepsy in the Era of Personalized Medicine was a comprehensive review of the factors that influence recommendations for genetic testing in epilepsy, the ILAE guidelines for the treatment of women with epilepsy during pregnancy, and personalized approaches to tumor-related epilepsy. It also highlighted specific pharmaceutical tools and medication management. Methods were employed to assess persons living with epilepsy for epilepsy surgery to maximize their seizure and nonseizure outcomes while avoiding patient bias. In summary, personalized medicine in epilepsy is an attainable goal with tremendous and lasting benefits.
Susan Spencer Symposium
Patient-Centered Research: Over a Decade of Impact on Practice
*Piero Perucca, MD, PhD, *Adam L. Hartman, MD, R. Edward Hogan, MD, Linda Huh, MD, Colin B. Josephson, MD, MSc, FRCPC, Alice D. Lam, MD, PhD, Daniel H. Lowenstein, MD, Page B. Pennell, MD
*Co-Chairs
This Symposium celebrated the 12th anniversary of the “Susan S. Spencer, MD Clinical Research Training Scholarship in Epilepsy” with a program highlighting the importance of patient-oriented research as well as opportunities and challenges in the field. The program started with reflections on the legacy of the late Dr Susan Spencer (Drs. Huh and Perucca), moving to showcase the research of 2 previous successful recipients of the Scholarship (Drs. Lam and Josephson), and concluding with a series of presentations and a multistakeholder panel discussion around strategies to foster the success of early career researchers (Drs. Lowenstein, Pennell, Hogan, and Hartman).
Reflections on Dr Susan Spencer’s Legacy
Drs. Huh and Perucca delivered complementary lectures on the legacy of Dr Susan Spencer (1948-2009), who was a Professor of Neurosurgery at Yale University School of Medicine and Co-Director of the Yale Epilepsy Program. Dr Spencer was a world authority in the field of epilepsy, in particular epilepsy surgery. She was firstly a brilliant and compassionate clinician, caring for thousands of patients who traveled from all over the world. She was a dedicated teacher and mentor, who trained dozens of epileptologists. Many of her former students/fellows have gone on to become research and/or clinical leaders in their own right. She was an outstanding researcher, with a strong track record in research funding. Her body of work, which includes >200 publications, has influenced clinical practice, research design/methodology, and modern concepts of epileptology. 125,126 She was also highly involved in the scientific community, holding several leadership roles (including serving as President of the American Epilepsy Society, AES, in 2000) and serving as a strong advocate for formal training in clinical research. In recognition of her legacy, the “Susan S. Spencer, MD Clinical Research Training Scholarship in Epilepsy” (https://www.aan.com/research/susan-s-spencer-md-clinical-training-scholarship) was established in 2009 to foster the development of investigators interested in pursuing careers in patient-oriented research.
Patient-oriented research along the epilepsy-dementia continuum
Dr Alice Lam, recipient of the 2017 Susan S. Spencer fellowship, described her path in building a patient-oriented research program at the interface between epilepsy and neurodegenerative diseases. Dr Lam shared the impact of the support she received from the Spencer fellowship, which came at a vulnerable point in her career as she was transitioning from epilepsy fellowship to a faculty position, and which allowed her to secure the time and resources needed to launch her research program. She described her research as being inspired by first-hand clinical observations of patients who presented with illnesses at the boundary between epilepsy and neurodegenerative diseases and who shone a light on our lack of knowledge in this area. She highlighted recent work from her lab, including the development of noninvasive, computational methods to detect hippocampal epileptiform activity 127 in patients with Alzheimer disease; and use of amyloid and tau PET imaging to examine mechanisms underlying the development of epilepsy in Alzheimer disease.
An odyssey from registries to EMR to high-dimensional data for patient-oriented research
Dr Josephson, recipient of the 2014 Susan S. Spencer fellowship, highlighted how the intersection of clinical registries, electronic and administrative health records (EMR/AHR) “Big Data,” and advanced analytics have catalyzed patient-centered research in epilepsy. Examples include the application of machine learning to patient-reported outcome measures (PROMs) and experiences (PREMs) data to identify unique clusters of psychosocial health, as well as clinical and socioeconomic features that predict group placement. 128 Likewise, EMR/AHR data have been leveraged to produce tools predicting levetiracetam-associated psychiatric adverse effects 129 and to show that mortality in epilepsy can be halved by referral to comprehensive epilepsy programs. 130 The availability of multimodal high-dimensional data comprising routine clinical, PROMs/PREMs, neurophysiological, neuroimaging, genetic, and pathological data has galvanized efforts to select the right treatment for the right patient at the right time, detect those at risk from specific comorbidities, and apply targeted interventions optimizing psychosocial health.
The role of an institution in creating a vibrant training program
Dr Lowenstein explained that, because the mission of universities is to generate new knowledge and train the next generation, academic medical centers have a clear responsibility to provide effective training programs for clinician-scientists across a broad range of scientific domains. Campus leadership works with leaders at the division, department, school, and other program levels to partner in providing these educational opportunities. Observations from former trainees who reflect back on their own experience emphasize the following issues or needs: finding a balance between research/clinical work/family/administrative tasks; difficulties in obtaining grants/proposing research as a clinician-scientist; economic disincentives to pursuing this pathway; exposures to research; fragile early career environment; uncertainty as to the best pathway; and mentoring. 131 Browsing the internet with terms such as “clinician-scientist training programs” leads to descriptions of many programs throughout the United States that are designed to address such issues and needs, with activities that include didactic learning, hands-on projects, works-in-progress sessions, assistance in grant writing, and mentoring/advising. In addition, institutions that have received Clinical-Translational Science Award (CTSA) grants from the National Institutes of Health (NIH) have benefitted from the priority placed on this type of training when the funding program was instituted in 2006. 132 The bottom line is there are many excellent programs in existence that may help junior investigators attain their goals (some of these programs are offered online or are available across institutions).
The role of a mentor
Dr Pennell emphasized that effective mentorship is essential to developing the next generation of patient-oriented researchers in epilepsy. Physicians are pivotal to clinical research, given their rare position to identify the critical questions from the clinical setting. Yet, time is limited during medical school and clinical training, with little exposure to the process of patient-oriented research. Obtaining research funding requires proficiency in fundamental skills to be competitive. Effective mentorship fills in these gaps. Studies of formal mentoring programs at individual academic medical centers demonstrate a 3- to 25-fold increase in the number of grants and total dollars awarded. 133 -135
Mentorship is a developmental partnership through which one shares knowledge, skills, information, and perspective to foster the personal and professional growth of others. A mentoring team is most effective, with multiple mentors playing different roles. Responsibilities of both the mentor and mentee include frequent and transparent communication, a mentorship agreement with goal setting, feedback, reevaluation, and adjustment. The responsibilities of the mentee also include giving back, thus passing on the benefits of mentoring to the next group of potential patient-oriented researchers.
The role of a professional society
Dr Hogan outlined how the American Epilepsy Society (AES) understands the importance of welcoming and involving early career clinical investigators. American Epilepsy Society provides early career investigators with broad exposure to many different colleagues with diverse expertise, which is especially important given a large number of causes, clinical manifestations, and comorbidities in patients with epilepsy. From its founding in 1936 as the “American League Against Epilepsy” with 69 members, 136 the AES now has >4000 members. The AES annual meeting provides a venue for the community of members with diverse interests to meaningfully interact. The AES Fellows Program involves 105 fellows who receive complementary annual meeting attendance, with special career development and mentoring programming. Finally, the AES provided nearly US$1.3 million for research grant funding in 2022, 87% of which was for early career grants. Therefore, AES provides a collegial environment, mentorship, and funding that is vital for our early career clinical investigators.
What is next after early career funding
Dr Adam Hartman discussed funding opportunities for investigators who have completed studies supported by early career foundation funding (e.g., the Susan Spencer Scholarship). The National Institute of Neurological Disorders and Stroke (NINDS) and other Institutes and Centers at the NIH support a variety of career development awards for junior faculty. Funding mechanisms include the K-series (e.g., K01, K08, K22, K23) but NINDS also supports applicants from diverse and underrepresented backgrounds through additional funding mechanisms (e.g., K01, K22). Other grants support those who are transitioning to independence (e.g., K02, K99/R00). National Institute of Neurological Disorders and Stroke provides diversity supplements in the form of short- and long-term investigator research supplements. Some private foundations and patient advocacy groups provide similar support. The NIH Lasker Clinical Scholars program aims to increase the number of clinical and translational researchers through a combination of intramural (NIH) and extramural mechanisms. Loan repayment programs also are available through NIH.
Additional information on NINDS Diversity programs can be found here: https://www.ninds.nih.gov/sites/default/files/documents/all_training_grants_handout_sept_2021_508c.pdf
The Susan Spencer Symposium highlighted some of the successes of foundation funding in the development of early career clinician-investigators. Additional support from academia, mentors of varied backgrounds, professional societies, and a broad spectrum of funders is available and can aid with the transition to independence.
Merritt-Putnam Symposium
Recent Insights Into Epileptic Networks and Clinical Implications
*Mohamad A. Mikati, MD, Fabrice Bartolomei MD, PhD, Ezequiel Gleichgerrcht, MD, PhD, Esther Krook-Magnuson, PhD, Jeffrey Noebels, MD, PhD
*Chair
Epilepsy is clearly a network disease. The past 2 years have witnessed major advances in the understanding of the underlying mechanisms of brain networks in epilepsy and their clinical implications. These advances span the spectrum from cellular, molecular, and animal model studies to connectome studies using novel artificial intelligence analysis methods and cutting-edge neuroradiological and neurophysiological data. The goals of this symposium were to (1) present the very recent advances in cellular, molecular, and animal model neuroscience that are elucidating these mechanisms; (2) review the recent studies in patients with various types of epilepsy that have clarified the pathophysiology of various networks in these patients and discuss their implications on improving the diagnosis, prognostication, and clinical management of patients with epilepsy.
Understanding neuronal synchronization in epilepsy: The impact of rare genetic mechanisms
Gene discovery is rapidly transforming the diagnostic and treatment landscape of epilepsy, moving the goalposts from seizure prevention to cure of many individually rare disorders. Each new gene raises the question of whether or how isolating rare monogenic disease mechanisms will benefit a far larger patient population. Analyses of these genes in mouse models reveal which circuits are unstable and when they can be rescued in the developing brain. 137 Emerging insights indicate that single gene defects lead to complex, spatially overlapping cellular excitability patterns, and these degenerate pathways offer multiple circuit solutions, expanding the number of therapeutic targets far beyond the mutated gene. The models confirm an important new axiom, that a single therapy may treat many genes, and for every gene there are many therapies. Nevertheless, recent evidence also suggests that simply restoring defective gene function may be a viable approach to reversing some developmental epileptic encephalopathies, 138 and at least one historical example suggests the next drug for an ultrarare monogenic epilepsy could become tomorrow’s blockbuster.
Epileptic neuronal circuits in epilepsy animal models: It is not just the neocortex
While the cerebellum is not traditionally considered part of seizure networks, substantial work has proven the old adage “the cerebellum is the one brain region that does not seize” categorically false. In fact, the cerebellum is engaged during a range of seizure types, including seizures with no overt behavioral component, and in some circumstances can even be the source of seizures. 139 The cerebellum is also a potential therapeutic target for a range of seizure types. For example, recent animal work utilizing online optimization methods indicates that electrical stimulation of the cerebellar cortex can reliably inhibit hippocampal seizures. 140 This provides hope that a clinical strategy targeting the cerebellum may be successful, although additional preclinical work is necessary. Expanding our view of epilepsy and seizure networks to include areas like the cerebellum can provide necessary insight into epilepsies and, importantly, potential new intervention targets and strategies.
Identification of human epilepsy networks and applications in epilepsy surgery
Human focal epilepsies are network diseases. Direct evidence came first from the quantification of SEEG recordings showing that the cerebral regions involved in seizure genesis are the seat of specific functional interactions. 141 The term “Epileptogenic networks” has been proposed to define networks involved in seizure generation or seizure propagation. 141 Quantified methods based on macroscopic brain modeling are currently being developed, in particular, the Virtual Epileptic Patient (VEP). 142 Virtual Epileptic Patient works by integrating the anatomical data of a patient (cortical parcellation, connectome) and a computational model of neuronal activity. This approach provides an estimate of the brain regions capable of generating seizures and is currently undergoing clinical evaluation in France (EPINOV trial). These approaches also offer new perspectives to simulate the effects of surgery (in silico) and a way to prognosticate at the individual level the results of surgery.
Artificial intelligence to characterize human epilepsy networks: Implications on classification prognostication
Dr Gleichgerrcht’s talk focused on the ways Artificial Intelligence (AI) can improve clinical care for patients with epilepsy. Using a real patient example, he showed how AI-based modeling can identify temporal lobe epilepsy from MRI scans. 143 His team is currently working on refining this technology to differentiate TLE from other conditions with shared radiographic findings, such as Alzheimer disease (i.e., medial temporal atrophy), and to determine the side of the brain where seizures originate. Dr Gleichgerrcht also discussed the potential of AI to predict surgical outcomes for epilepsy patients, with a focus on forecasting which patients will become seizure-free after surgery based on preoperative scans. 144 Overall, his talk emphasized the value of AI as a tool for clinicians treating epilepsy patients.
Conclusions
The study of the pathophysiology of epilepsy and the development of novel therapies are being enhanced by novel genetic, neurophysiological multimodality, and artificial intelligence methods that address the network aspects of its pathophysiology.
Monogenic epilepsy therapy discovery extends beyond mutation carriers as it uncovers epilepsy and anti-seizure mechanisms that apply to other patients.
The cerebellum modulates focal and generalized seizure circuits and its stimulation can abort these seizures in animals raising the potential for human applications.
Computational neuroscience modeling of epileptic networks is offering additional tools to guide surgical resections.
Hub mapping and AI techniques can help predict clinical trajectories and assist in clinical decisions.
Special Lecture
Sleep and Epilepsy Across the Life Span
*Gordon F. Buchanan, MD, PhD, *Milena K. Pavlova, MD, Gita Gupta, MD, Sanjeev V. Kothare, MD, Alice D. Lam, MD, PhD, MS, Mark Quigg, MD, MSc, Renée A. Shellhaas, MD, MS
*Co-Chairs
Sleep, circadian rhythms, and epilepsy interact in profound ways. While this may appear intuitively obvious, it is easy to forget that the presentation of sleep disorders, sleep needs, and circadian rhythms differ among individuals and change with age. Indeed, the interplay among sleep, circadian, rhythms, and epilepsy evolves throughout the developmental stages of the life span continuum. The goal of this Special Lecture, organized by the Sleep and Epilepsy Workgroup of the American Epilepsy Society, was to discuss the evolution of sleep and epilepsy across the life span.
Epilepsy is a common neurological disorder affecting about 50 million people worldwide. 145 It is well appreciated that there is a bidirectional relationship between sleep and epilepsy. 146 What is less well-appreciated is how much sleep and epilepsy change throughout the life span, and how the relationship between them also changes. 147,148 For this Special Lecture at AES 2022, experts in sleep and epilepsy from each domain along the life span continuum were assembled to discuss how sleep and epilepsy change across the life span from the neonatal period and infancy (Renée Shellhaas), progressing through childhood (Sanjeev Kothare), then adolescence (Mark Quigg), adulthood (Milena Pavlova), and older adulthood (Alice Lam). We also explored the impact on caregivers’ sleep of caring for people, especially children, with epilepsy (Gita Gupta). The session was introduced by Gordon Buchanan. Each talk was framed by a clinical case that represented a set of age-specific clinical questions and continued to a discussion of the basic science mechanisms and clinical relevance of these questions. This was a lively and well-received session, with an active question and answer panel at the end moderated by Drs. Pavlova and Buchanan. Each talk is summarized below.
Sleep in neonates and infants with seizures
Sleep is a fundamental biomarker of brain function in newborn infants and is a primary concern for parents. The presence of sleep–wake cycling is a good prognostic indicator for neonates who require intensive care. 149 -151 The quality of nonrapid eye movement (NREM) sleep, including low-frequency EEG power and brain oxygen extraction in this stage, is predictive of 18-month neurodevelopmental outcomes. 152 Improving sleep physiology holds promise as an intervention that could lead to improved neurodevelopment.
Safe sleep practices (babies should be placed on their back to sleep, alone in their crib, with a firm mattress and no bedding, toys, or other objects) can save lives by preventing sudden infant death syndrome. Modeling safe sleep behaviors in the hospital setting is key. 153 Other opportunities to optimize infant sleep in the hospital setting include avoidance of hands-on care (particularly during rapid eye movement [REM] sleep), attention to light/dark cycles, and appropriate exposure to spoken language with avoidance of excessive noise. 154
Sleep and epilepsy in the pediatric population
Pediatric epileptologists should be aware of the age-related sleep requirements across the lifespan, especially the changes in nap time and duration in young children. 155 The etiology of sleep disruption in patients with epilepsy is multifactorial and includes factors such as inadequate sleep hygiene, coexisting sleep disorders, circadian rhythm disturbances, epilepsy per se, seizure frequency, and the effect of antiepileptic medications. 156 Fragmented sleep causes daytime fatigue and sleepiness along with poor seizure control, while uncontrolled seizures and epilepsy can worsen sleep quality. Improving either or both can improve both sleep and epilepsy and overall improve quality of life (QOL). Sleep hygiene and sleep schedules can go a long way in consolidating nocturnal sleep.
There is a distinct circadian pattern to interictal epileptiform activity and seizures 157 in the pediatric population. Interictal epileptiform activity and seizures are more likely during NREM sleep and rare during REM sleep. 158 Certain pediatric epilepsy syndromes like infantile spasms, benign rolandic epilepsy, benign occipital lobe epilepsy, juvenile myoclonic epilepsy, and frontal lobe epilepsy have a distinct sleep–wake signature is well established. The marked activation of epileptiform activity during sleep in conditions such as electrical status epilepticus in slow wave sleep (ESES) results in cognitive impairment due to disruption of sleep-related synaptic reorganization. 159
A large proportion of patients with epilepsy have comorbid obstructive sleep apnea (OSA). In addition, vagal nerve stimulation is known to worsen OSA. 160 Treatment of that will not only improve sleep disruption but also improve seizure control and improve daytime alertness.
Sleep and epilepsy across the life span: Adolescence
The developing adolescent brain is caught between the childhood need for sleep time (>9 hours), a shift from an early to late preferred phase of activity in the circadian “day,” and rising social pressures and autonomy, all of which render adolescents and young adults susceptible to sleep deprivation. 161 Sleep deprivation, in turn, contributes to the activation of interictal epileptiform discharges and to the precipitation of seizures, especially in certain epilepsies such as juvenile myoclonic epilepsy. 162 Chronic insomnia affects more than a third of patients with epilepsy, and rising evidence shows that sleep hygiene and other measures to treat insomnia can improve seizure control and improve quality of life. 163 The temporal patterns of interictal discharges and seizure occurrence are the cumulative effect of sleep–wake state, sleep deprivation, the circadian timing system, and other multidian endogenous and exogenous factors; isolating the seizure-promoting and inhibiting aspects of these entwined rhythms makes explorations of mechanisms difficult. 164 The circadian timing system also affects the metabolism of certain anti-seizure medications, and evening-weighting of medications may help limit toxicities and improve seizure control. 165 Use of a simple set of sleep screening questions, as well as broadly applicable advice for organizing daily activity to promote sleep health, can improve sleep, which, in turn, can help convert those with continuing or breakthrough seizures into those with improved seizure control. 166
Sleep and epilepsy interactions in the adult
In adulthood, sleep architecture continues to evolve, albeit more slowly than in younger years. The overall proportion of REM sleep decreases from approximately 25% to ∼15%. Slow-wave sleep decreases from ∼22% to near none in later adulthood. The circadian cycle affects both frequency of epileptiform discharges as well as sleep stage. In healthy individuals, as well as in epilepsy, REM sleep is heavily regulated by the circadian cycle, peaking after the body temperature nadir. In contrast, slow-wave sleep has a predominantly homeostatic control and increases proportionally after sleep deprivation.
Seizure frequency is highest in NREM stage N2 sleep (44% of all seizures), while less than 1% of seizures occur in REM. The mechanisms for this stage predominance remain to be elucidated. 6
Common sleep disorders in the adult include insomnia, circadian rhythm disorders, restless legs syndrome, and OSA. Rarer disorders include narcolepsy and other central hypersomnias. Evaluations may include a standardized sleep log, actigraphy, polysomnography with or without extended EEG, and measurements of dim light melatonin onset time.
Treatment of comorbid sleep disorders can lead to improved seizure control. For example, in a recent study patients with epilepsy and comorbid OSA treated with positive airway pressure (PAP) had reduced seizure frequency even without a change in anti-seizure medications. 167 However, adherence to PAP therapy can be challenging for epilepsy patients, 168 especially in the first months of therapy.
Sleep and epilepsy in older adults
Adults with epilepsy are 2-3 times more likely to develop sleep disorders compared to the general population, including insomnia, OSA, and restless leg syndrome. 169 Changes in sleep also occur with aging, including reduced total sleep time, reduced slow wave sleep, increased sleep fragmentation, and increased prevalence of OSA. 170 Bidirectional relationships exist between epilepsy, dementia, and stroke, and older adults with epilepsy have a 2-3 times increased risk of developing incident dementia and/or stroke compared to age-matched controls. Management of epilepsy in older adults should therefore include assessment and reduction of risk factors for dementia and stroke. 171 -173 Sleep disorders, including short sleep time, OSA, and disrupted slow-wave sleep, represent highly prevalent and shared risk factors for epilepsy, dementia, and stroke. Diagnosis and treatment of these sleep disorders may be a promising approach to improve seizure control and reduce dementia and stroke risk in older adults with epilepsy. 174,175 Studies evaluating the clinical impact of sleep disorders in older adults with epilepsy are needed.
Effects of epilepsy on caregivers’ sleep
Caregivers of children with epilepsy are at risk for insufficient sleep (<7 hours total sleep time) compared to both caregivers of children with other chronic illnesses and caregivers of typically developing children. On average, they may get 4.5 hours of sleep. 176 They also experience more parenting stress compared to caregivers of children with other neurologic conditions. 177 Factors such as nocturnal seizure monitoring, room sharing, and cosleeping contribute to chronic caregiver sleep deprivation. 178 Consequences of chronic sleep deprivation may include difficulty with providing complex medical care, 179 chronic illness such as diabetes and cardiovascular disease, 180 increased mental health burdens, 181 and adverse financial and social effects. Clinicians should ask about sleep problems of the child–caregiver dyad and feel comfortable referring if appropriate. 182 Lastly, advocacy for sustainable access to home respite care may promote adequate sleep for caregivers of persons with epilepsy.
Epilepsies and sleep–wake regulation change considerably throughout the life span, as do circadian and other biological rhythms. Sleep and circadian phase influence seizures and epilepsy. Seizures and epilepsy influence sleep and circadian regulation. These interactions also evolve throughout the lifespan. Disordered sleep associated with epilepsy greatly increases comorbidity. Certain sleep disorders can be particularly challenging to manage in persons with epilepsy. Sleep disorders and epilepsy can be difficult to identify in certain populations, such as in patients with new-onset dementia. When considering the impact of impaired sleep and circadian rhythms in persons with epilepsy, it is important to also consider the impact of epilepsy on sleep and circadian health in their caregivers. This is an interesting and complex field with interactions at many levels. A better understanding of the interactions will continue to emerge in the coming years and is sure to improve management and quality of life for those with epilepsy.
Pediatric State of the Art Symposium
Addressing Knowledge Gaps in Early Life Epilepsy
*M. Scott Perry, MD, Zachary Grinspan, MD, MS, Michael Hammer, PhD, Kerri L. Neville, MD, Chima O. Oluigbo, MD, Daniel W. Shrey, MD, FACNS, Rani K. Singh, MD, Saher Suleman, MD, Elaine Wirrell, MD
*Chair.
The incidence of childhood epilepsy is highest in the first years of life. Genetic and structural etiologies abound in this age group and resistance to anti-seizure medications (ASM) is common, leading to significant adverse consequences for cognitive and behavioral development. The Agency for Healthcare Research and Quality recently highlighted significant knowledge gaps for the treatment of early life epilepsies, including the absence of data for the effectiveness of currently available medications, the effectiveness of nonpharmacologic treatments, and the harms of these treatments used early in life. 183 This symposium highlighted multiple innovative and collaborative efforts to broaden research into early life epilepsies in an effort to eliminate knowledge gaps and improve outcomes.
Lessons learned through early victories: The early life epilepsy registry
The Early Life Epilepsy study was designed to evaluate the range of epilepsy type(s), etiologies, investigations and their yield, treatments prescribed, and their efficacy and outcomes for children with onset of epilepsy prior to 36 months of age. Patients were recruited prospectively at 17 US centers over a 3-year period and charts were reviewed at baseline and every 3 months through the first year after epilepsy diagnosis.
Regarding investigations, both neuroimaging, most commonly performed with epilepsy protocol MRI, and genetic studies (including chromosomal microarray, epilepsy gene panel, and whole exome sequencing among others) were high yield, documenting an underlying etiology in approximately 40% of cases each. 184,185 Importantly, of children who had abnormal imaging, 44% were also found to have pathogenic gene variants, illustrating that the underlying etiology is often structural-genetic, as opposed to structural or genetic. 185 Abnormal imaging was more commonly seen with earlier onset of epilepsy, associated developmental delay, epileptic spasms, and focal or unclear onset seizures. 185
Of those with epilepsy onset before 12 months, 42% presented with spasms as their initial seizure type and an additional 8% evolved to spasms over the course of their epilepsy. 186 Gestational age was negatively correlated with the age of spasm onset suggesting that the infant brain must achieve a certain postconceptual age to manifest spasms. Spasms were preferentially associated with broad developmental and regulatory pathways or etiologies that impacted neuronal cell body organelles, whereas pathways affecting cell motility; stimuli and ion channels; and axonal, dendritic, and synaptic regions were more commonly associated with nonspasm seizures.
Regarding the short-term outcome of early life epilepsy, the mortality rate was 2.9% in the first year; however, no death occurred in children with unknown etiology and normal development. 187 Drug resistance was seen in 35% of cases with significant predictors being at age of onset before 12 months and developmental delay. 187 In the presence of both of these factors, 54% of children were drug resistant and in their absence only 21% were drug resistant. Approximately 15% of infants presenting with other seizure types ultimately evolved to infantile spasms and such evolution was significantly more likely if initial seizure onset was prior to 3 months of age and in the presence of developmental delay. 187 Nearly one quarter of children with typical or only equivocal delay at seizure onset progressed to definite delay after 1 year of follow-up. 187 Risk factors included equivocal or mild delay versus normal development at onset, onset prior to 12 months of age, known etiology and drug resistance. In the absence of any of these factors, less than 3% had developmental decline; however, in the presence of all 4 factors, 91% showed decline.
The pediatric epilepsy learning healthcare system (PELHS): Answering big questions with bigger data
Several key questions about the clinical approach to early life seizures and epilepsy merit attention from clinician scientists. These include understanding the value of early genetic testing, developing guidance to select the most efficacious first and second-line ASMs, and developing and validating early biomarkers to predict epilepsy evolution, relapse, or refractoriness.
Landmark studies from the past decade have begun to answer each of these questions. For example, the importance of genetic testing is well established, with yields as high as 80% in neonatal epilepsies. 188 High-quality evidence point to phenobarbital as the preferred first-line agent for neonatal seizures, ACTH/prednisolone and/or vigabatrin as first line for infantile spasms, sodium channel blockers for some specific genetic epilepsies like KCNQ2, and levetiracetam for other infantile onset epilepsies. 17,189 -192 The evidence for second-line therapy is emerging—as an example, for infantile spasms that do not respond to vigabatrin, hormonal therapy (ACTH/prednisolone) is preferred; and if hormonal therapy was used first, vigabatrin. 193 In neonates with acute symptomatic seizures, combinations of clinical biomarkers can predict infantile spasms with a positive predictive value above 50%. 194 In children with epilepsy due to cortical dysplasia, failure of a single medication to control seizures is a very strong herald of pharmacoresistance. 195
Much of the published evidence and planned research relies on techniques for causal inference using observational methods. These studies require careful attention to methodologic pitfalls, including treatment selection bias, clustered outcomes, missing data, and unmeasured confounders. Learning health care systems, which systematically collect electronic health record data from multiple centers and centralize those data at a data coordinating center, are an emerging source of data for this work. The pediatric epilepsy learning health care system (PELHS) and the epilepsy learning health care system (ELHS) are examples of these systems for people with epilepsy. 196 -198 Early work from PELHS includes the development of a novel quality of life measure for use in clinical care, and the development, iteration, and deployment of a standard electronic health record form optimized to support clinical workflows. As these data are collected, we anticipate an increasing pace of discovery to optimize clinical care for these vulnerable children.
What we don’t know can hurt others: Disparities in treatment of infantile spasms
The National Infantile Spasms Consortium, a collaborative, prospective, multicenter database, investigated important questions in disparities in the treatment of infantile spasms. 199 There were 25 centers from across the country and 555 infants with new-onset infantile spasms included. Children who identified as Black/non-Hispanic had a lower odds of receiving a standard treatment course for infantile spasms (P = .001). The treatment course was defined as standard therapy for both the first treatment, and, when needed, the second treatment. This remained statistically significant even after adjusting for a number of clinical variables such as etiology, history of prior seizures, or developmental delay. Children with public insurance were also less likely to receive standard therapy for their first treatment (P = .01). Action is needed to ensure all children are able to benefit from advances in the field of pediatric neurology. This could include creating protocol-driven treatment pathways, advocating with state Medicaid programs, identifying and resolving institutional health system barriers, and on an individual level, recognizing the potential role of implicit bias affecting treatment recommendations.
Strength in numbers: Developing biomarkers in rare epilepsies
The incidence of rare epilepsies often precludes successfully recruiting sufficient sample sizes at a single institution, especially for prospective studies. Conditions with heterogenous etiologies, such as infantile spasms, which have more than 300 known causes, 200 further complicate this process. Regional collaborations are beneficial for pilot data, proof-of-concept, and validation/reproduction of prior work. Multi-centered consortia-based research studies are integral for making clinical discoveries in a timely fashion. They support accelerated recruitment, pooled expertise and assets, sharing of data, and mentorship. Leveraging existing infrastructure for novel projects is recommended. Establishing a successful research record of accomplishment within a consortium is critical for obtaining funding, especially from federal entities. Disseminating practice-changing findings can be accomplished by leveraging learning health care systems. Future work should focus on modernizing data management strategies and developing cloud-based data solutions with autonomous deidentification and standardization of data. This would significantly lower the barriers to doing multicentered research.
Debate: Early surgery versus waiting for a second ASM failure—Con
Epilepsy surgery is accepted for the management of drug-resistant focal epilepsy that persists despite 2 adequate ASM trials. 201 However, expedited epilepsy surgery is not without risk. Although complication rates have decreased over the last > 30 years, complications remain an unavoidable consequence of epilepsy surgery and include the risk of bleeding, infection, and persistent neurologic deficits. Surgical treatment of epilepsy does not always stop seizures: especially in the setting of focal cortical dysplasia (FCD), in which the risk of repeat surgery ranges from 6% to 23%. 202 Expedited epilepsy surgery is a challenge to our conventional practice: even though earlier surgery may be associated with better seizure outcomes, it is only speculative that early surgery may limit brain network dysfunction, be cost-effective, and may lead to better seizure outcomes. 203
Debate: Early surgery versus waiting for a second ASM failure—Pro
The evidence basis for the efficacy of pediatric epilepsy surgery is well established. 204 However, the timing of surgery in early life epilepsy is not well defined. Traditionally, epilepsy surgery is considered when a patient has “medically refractory epilepsy” defined as: “Inadequate seizure control despite appropriate medical therapy with at least 2 ASMs in maximally tolerated doses for 18 months to 2 years, or adequate seizure control with unacceptable drug-related side effects.” 205 The rationale for this decision is that there is only a 5% to 10% probability of seizure control with a third drug. 206
However, this definition of ASM refractoriness is not appropriate in children with early life epilepsy because of the significant cognitive impact of early life epilepsy, even in a relatively short period of time. Sorg et al showed that there is an increase in an absolute and relative increase in cognitive impairment in toddlers compared to preschool and school children with uncontrolled epilepsy. 207 Freitag and Tuxhorn showed that gain or losses in cognitive function in preschool children after epilepsy surgery is proportional to the duration of epilepsy, thus establishing a rationale for early epilepsy surgery. 208
In studying the prevalence and risk factors associated with pharmacoresistance in children with FCD-related epilepsy, Cohen et al noted that failure of one ASM is associated with substantially increased risk of pharmacoresistance (odds ratio of 346). 195 The authors, therefore, recommended a redefinition of pharmacoresistance in FCD-related epilepsy to the failure of one ASM and proposed early epilepsy surgery in this patient population.
Early epilepsy surgery in early life epilepsy is effective in disrupting the complex interplay of factors such as the effect of uncontrolled epilepsy and ASM exposure which lead to altered structural and functional connectivity of neural networks and cognitive impairment. 209 Thus, effective early surgical intervention limits the duration of epilepsy which is the only modifiable predictor of maladaptive cognitive development in children with early life epilepsy. Finally, epilepsy surgery in the very young has been shown to be very safe and effective and therefore should be recommended promptly in early life epilepsy. 210
SCN8A registry
Pathogenic variants at the voltage-gated sodium channel gene, SCN8A, are associated with a wide spectrum of epilepsy phenotypes ranging from benign familial infantile seizures to mild-to-severe developmental and epileptic encephalopathies. Clinical features include mild to severe developmental impairment and multiple seizure types, including generalized tonic–clonic and absence seizures, focal seizures, and infantile/epileptic spasms. One of the major challenges facing clinicians and researchers is to identify genotype–phenotype correlations that may improve prognosis, guide treatment decisions, and ultimately lead to precision medicine approaches. The SCN8A Registry is a patient-driven online database collecting data on children with SCN8A-related disorders. The range of data types collected provides an opportunity to perform genotype–phenotype correlation analyses and to develop methods to classify patients based on clinical features associated with gene variants. We develop predictive modeling approaches to (1) classify patients carrying gain (GOF)- or loss-of-function (LOF) variants based on features present at initial diagnosis, and (2) subdivide patients with GOF variants into distinct severity subgroups. We find that patients with milder variants have a later age at seizure onset, higher rates of seizure freedom, higher developmental quotient, and require fewer ASMs to manage seizures.
Hot Topics Symposium
From Traumatic Brain Injury to Post-Traumatic Epilepsy and Its Comorbidities
*Asla Pitkänen, MD, PhD, *Ramon Diaz-Arastia, MD, PhD, Jeanne T. Paz, PhD, Eugen Trinka, MD, MSc, FRCP, Mary Jo Pugh, PhD, RN
*Co-Chairs
Approximately 70 million individuals are estimated to suffer traumatic brain injury (TBI) each year. Epidemiologic studies indicate that the risk of developing epilepsy (epileptogenesis) increases according to the severity of TBI, being about 2- to 4-fold after mild, 8-fold after moderate, and 16-fold after severe TBI. 211 Post-traumatic epilepsy (PTE) is estimated to account for approximately 5% of all epilepsies and 20% of structural epilepsies. Mild TBI comprises over 90% of all TBI, and thus the total number of patients developing epilepsy after mild TBI can be expected to be greater than that of patients developing epilepsy after severe TBI, which has been the focus of experimental and clinical PTE studies. However, the brain pathologies and the early postinjury factors, contributing to the risk of PTE after different TBI severities are incompletely understood. Despite favorable preclinical proof-of-concept treatment trials in models of PTE, no anti-epileptogenic or disease-modifying treatments are available in the clinic. The symposium presented the current state-of-art in understanding who is at risk of PTE and what are the treatment options now and in the future.
What have we learned on post-traumatic epilepsy in prospective large multicenter studies—Is there an epileptogenic TBI endophenotype?
Several recent epidemiologic studies have highlighted the risk of early and late post-traumatic seizures not only after a severe but also after a mild TBI. These studies also indicate the heterogeneity of impact types and consequent pathologies that can lead to epilepsy, implying mechanistic heterogeneity of post-TBI epileptogenesis. Ongoing prospective observational studies such as EpiBioS4Rx (www.epibios.loni.usc.edu/) and TRACK-TBI EPI (www.tracktbinet.ucsf.edu/track-tbi-epi) focus on the discovery of physiologic, neuroimaging and molecular biomarkers for post-traumatic epileptogenesis. These studies have already revealed that neuroimaging of PTE is often nonlesional, reflecting diffuse and microscopic injury. Several open questions remain. How often is PTE effectively controlled by anti-seizure medications? How are the TBI comorbidities (cognitive and affective disorders, functional limitations) affected by late post-traumatic seizures? For nonlesional PTE, what are the associations with traumatic axonal and/or microvascular injury and with molecular biomarkers of neuroinflammation?
Sleep, inflammation, and inhibitory microcircuits—Is thalamus emerging as an epicenter for post-traumatic epileptogenesis?
Traumatic brain injury can have long-term consequences, including cognitive dysfunction, disruptions to sleep, and post-traumatic epilepsy. Although the initial injury impacts the cortex, these long-term disabilities are due to secondary damage that accrues over time, particularly in the thalamus, which has reciprocal connections with the cortex. In a mouse model of mild TBI (mTBI), we found that levels of the immune molecule C1q were increased in the corticothalamic system, specifically at sites of neuron loss and inflammation. This increase correlated with disruption in sleep spindles and the emergence of epileptic activities. Remarkably, blocking C1q counteracted these outcomes, pinpointing C1q as a potential therapeutic target for mTBI. 212 Furthermore, we discovered that triggering thalamic neuroinflammation in mice was sufficient to induce thalamocortical hyperexcitability and seizure risk, even in the absence of traumatic injury. These data indicate that inflammation in the thalamus is a promising target for treating TBI-related disabilities. 213
Prophylaxis and/or treatment of acute post-TBI seizures and status epilepticus: Who, when, why, which, and how—Are there enough data for consensus?
At least 2% to 15% of patients with moderate to severe traumatic brain injury (TBI) will suffer from acute symptomatic seizures. 214 Most studies report figures between 2% and 5%. Half of these acute symptomatic seizures occur in the first 24 hours and rates of status epilepticus (SE) vary between 0.2% and 5%. Acute symptomatic seizures in TBI are associated with increased mortality, an increased risk for the development of posttraumatic epilepsy, and with a poor functional outcome.
There is more than 50 years of clinical research in preventing acute symptomatic seizures and its sequelae, especially posttraumatic epilepsy (PTE). With our currently available anti-seizure medicine, we can significantly lower the risk of early posttraumatic seizures occurring in the first 7 days after injury, but these treatments have no significant effect on the risk of late posttraumatic seizures. 215 Unfortunately, anti-seizure medicines are not without any harm, especially on neurocognitive and rehabilitation outcomes, independent of the onset of epilepsy.
There is significant clinical variability in the current practices of pharmacological management of acute posttraumatic seizures in adults across the globe.
Despite major advances in epidemiology and pathophysiology, some questions still remain unanswered: What is the significance of small posttraumatic MRI abnormalities? What is the significance of ictal EEG abnormalities and nonconvulsive seizures and SE? Are mild TBIs at all associated with epilepsy? What is the influence of anti-seizure medicines on recovery and long-term outcomes? Only sound clinical trials can answer these questions and there is enough evidence to design such.
Can post-traumatic epileptogenesis be prevented—Any hope on the horizon?
Several animal models of TBI produced in mice, rats, or pigs used to show increased seizure susceptibility or epilepsy in chronic electroencephalogram (EEG) follow-up. Thus, we now have clinically translatable models, representing different injury mechanisms to assess the multiple mechanisms and discover biomarkers and antiepileptogenic treatments for a heterogeneous group of patients with PTE. So far, almost 20 molecular, imaging or electrophysiological biomarkers have shown promise as prognostic biomarkers for post-TBI epileptogenesis. 216 Use of shortening of sleep spindles as diagnostic biomarkers to identify subjects undergoing epileptogenesis and pinpointing the epileptogenic region by using high-frequency oscillations as diagnostic biomarkers of epileptogenic tissue provide hope for pathology-specific antiepileptogenic drug development. So far, more than 20 therapies, including small molecules with different mechanisms of actions and one-cell therapy approach have shown promise as antiepileptogenic agents. 216 To speed up the progress there is a need to expand the use of well-characterized reproducible PTE models in multicenter design to guarantee statistical power in clinically relevant preclinical studies. The use of emerging biomarkers for the identification of subjects at risk and delineation of the epileptogenic zone open avenues for sophisticated systems biology approaches in profiling of the epileptogenic region for target identification. There is hope on the horizon.
Quality-of-life determinants in post-traumatic epilepsy: Is it just about the seizures? Quality of Life is an important outcome for people with epilepsy
Quality of life (QoL) in epilepsy is a critical indicator of patient well-being. Meta-analysis identified energy/fatigue and seizure frequency as the most important predictors of epilepsy QoL followed by comorbidity. 217 Recently this inquiry expanded to traumatic brain injury (TBI) and posttraumatic epilepsy (PTE). US military Veterans with PTE had significantly lower scores on diverse measures of QoL (epilepsy/TBI specific, generic) compared to those with epilepsy alone. Those with PTE were more likely to have drug-resistant epilepsy (DRE; 44%) versus epilepsy (33% P < .01); those with PTE-DRE had the lowest QoL scores (P < .05). 218 Further analysis revealed that seizure-related variables accounted for the most variance in epilepsy-specific and physical QoL measures; psychiatric comorbidity was most important for TBI and mental health–focused QoL measures. Sleep and pain accounted for significant variance for Physical QoL. These findings suggest that QoL is multimodal and holistic approaches to assessment and treatment are needed to optimize patient outcomes.
Scientific Symposium
The Many Facets of Neurodegeneration in Epilepsy
*Andrea Bernasconi, MD, Boris Bernhardt, PhD, Jeannie Chin, PhD, Matthias Koepp, MD, PhD, Carrie McDonald, PhD, Maria Thom, MD, MBBS
*Chair
Characterizing neurodegenerative pathology in epilepsy and potential drivers
The pathological identification of neurodegenerative disease processes in patients undergoing epilepsy surgery is relevant to understanding the causes of progressive regional brain atrophy reported in imaging studies and cognitive deficits. 219 Several studies have investigated the prevalence of Alzheimer type pathology (tau protein/beta amyloid) in surgical pathologies including mTORopathies, as focal cortical dysplasia. Questions remain regarding (i) drivers for tau-phosphorylation in epilepsy (e.g., seizure activity, mTOR activation, genetic factors), its reversibility, and/or if there is progressive accumulation. In postmortem epilepsy studies, relationships between traumatic brain injuries and tau accumulation, with patterns of chronic–traumatic encephalopathy, have been recognized. Neuroinflammation in the setting of seizures may also promote tau phosphorylation and microglial upregulation has also been implicated as a modifying factor for regional gray matter thinning. 220 Finally, white matter pathology and microangiopathy, as in vascular dementias, represent a less explored but further degenerative process in epilepsy of potential clinical relevance.
Shared mechanisms of cognitive decline in Alzheimer disease and epilepsy
Seizures are associated with cognitive deficits, but how they impair cognition is unclear, particularly for conditions in which seizures are relatively infrequent, such as Alzheimer disease (AD). Our work in mouse models demonstrated that seizures induce activity-dependent expression of ΔFosB, 221 a transcription factor with an unusually long half-life, that epigenetically suppresses expression of genes necessary for plasticity and memory. The magnitude of ΔFosB expression directly corresponds with poorer cognitive scores in both mice and patients with AD. Short-term blockade of ΔFosB derepresses gene expression and improves hippocampal memory, indicating that ΔFosB is critical for seizure-induced memory deficits. Since aberrant ΔFosB expression occurs in individuals with epilepsy, patients with AD, and in mouse models of either condition, it is likely relevant to any condition with seizures. 222 Chromatin–immunoprecipitation sequencing studies indicate that ΔFosB regulates several domains of neuronal function and reveal how even infrequent seizures lead to persistent changes in neuronal function.
Cognitive profiles associated with neurodegeneration in epilepsy
Cognitive profiles in older adults with epilepsy suggest that up to 60% meet the criteria for a cognitive disorder of aging (e.g., mild cognitive impairment; MCI). 223 Several risk factors that may contribute to the co-occurrence of MCI, age-accelerated cognitive decline, and progression to dementia in epilepsy have recently been identified. These include APOE genotype, hypertension, diabetes, and heart disease as risk factors for global cognitive decline, 224 as well as higher education as a buffer against cognitive decline. A critical limitation is that most studies have utilized cognitive screenings, which often lack sensitivity for detecting MCI and longitudinal changes in cognition. Our data support the use of comprehensive neuropsychological testing in older adults that enable a better characterization of cognitive phenotypes and epilepsy/MCI subtypes. Cognitive phenotyping, when combined with other biomarkers, may lead to earlier detection of patients at risk for progression to dementia and a more accurate classification of dementia etiology.
Imaging correlates of pathological ageing in epilepsy
MRI studies show epilepsy syndrome-specific patterns of progressive regional GM atrophy beyond normal aging, implying disease-related effects. These patterns differ from AD and chronic traumatic encephalopathy (CTE). Proposed mechanisms include accelerated normal aging, blood–brain–barrier dysfunction, altered excitability with seizure-network/neuronal activity-dependent degenerative alterations, cumulative minor traumatic head injuries, and degenerative vascular pathology, all of which may act synergistically. Notably, PET-tau tracers can detect pathology in AD prior to cognitive decline and predict the disease course. In epilepsy, 18F-MK6240 tracer showed tau increases comparable to reductions in cortical thickness on MRI. Neuronal and glial tau pathology shows features of both AD and CTE in addition to unique patterns that could be seizure-specific, suggesting mixed etiological neurodegenerative mechanisms in epilepsy. 225 Thus, AD and epilepsy are unlikely to be coincidental pathologies. In a subgroup of patients, epilepsy is a progressive disease, characterized by tau pathology that could be reversible or activity dependent.
Multimodal connectome models of epilepsy-related disease progression
Neuroimaging indicates that drug-resistant temporal lobe epilepsy (TLE) is associated with progressive GM atrophy that is distinct from healthy aging, with effects in mesiotemporal, subcortical, and neocortical structures, often bilateral. 226 Progression may vary, with older patients and those with frequent seizures being more at risk. Cross-referencing epilepsy-related atrophy patterns with connectome models suggests increased susceptibility of densely-connected hubs, and identifies temporo-limbic disease epicenters. Expanding from ex vivo findings showing hyperphosphorylated tau in surgical tissue, novel positron emission tomography tracers have imaged tau deposits in vivo. Preliminary data suggest that TLE patients indeed show increased tau levels compared to controls in a widespread and bilateral distribution. 227 Findings correlate with epilepsy duration and measures of seizure burden, but associations to cognitive dysfunction appear complex. Longitudinal studies that enroll patients at various disease stages, and with variable levels of drug control, will clarify the causes and consequences of epilepsy, and identify micro- and macroscale mechanisms contributing to disease progression.
Footnotes
Authors’ Note
This report does not represent the official view of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institutes of Health (NIH), or any part of the US Federal Government. No official support or endorsement of this article by the NINDS or NIH is intended or should be inferred.
Acknowledgments
The authors would like to thank the AES staff including Eileen Murray, MM, CAE, Shawna Strickland, PhD, CAE, RRT, FAARC, Susan Oliver, MBA, Cristina Graham, JoLynn Amsden, Amy Kephart, MPH, CAE for their support and all the committee members in charge of program planning of the Annual American Epilepsy Society Meeting.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.