
Abstract
Whether genetic factors contribute to acquired epilepsies has long been controversial. Supporters observe that, among individuals exposed to seemingly the same brain insult, only a minority develops unprovoked seizures. Yet, only in relatively recent years have studies started to build a case for genetic contributions. Here, we appraise this emerging evidence, by providing a critical review of studies published in the field.
Introduction
Epilepsy due to an acquired brain insult, such as a traumatic brain injury, stroke, tumor or central nervous system (CNS) infection (collectively termed “acquired epilepsy”), accounts for approximately one-third of all epilepsies. 1 Importantly, only a subset of individuals who sustain a brain insult develop epilepsy, for example, 2% to 5% of those sustaining head trauma, 2% to 4% of those having a stroke, and 5% to 10% of those who have a CNS infection. 2,3 The risk of acquired epilepsy depends on the characteristics of the brain insult, for example, type, severity, size, and location as well as factors related to the individual, for example, age, sex, and genetic factors. 3 In this setting, genetic factors do not affect the epilepsy risk directly, but they operate by increasing susceptibility to the consequences of the acquired brain insult.
A genetically determined vulnerability to acquired epilepsy has long been postulated and debated. Initial evidence stemmed from the clinical observation that among people who seemingly had the same brain insult, some develop unprovoked recurrent seizures and others do not. 4 However, data supporting such vulnerability have remained largely elusive until recent times. Only in the past decade or so have studies started to shed some light on the genetic contributions to specific forms of acquired epilepsy. Here, we review this recent body of work, which largely relates to 3 types of acquired epilepsy: post-traumatic epilepsy, poststroke epilepsy, and temporal lobe epilepsy with hippocampal sclerosis.
Post-Traumatic Epilepsy
Traumatic brain injury is an important cause of acquired epilepsy, accounting for 15% of cases and for about 30% of acquired epilepsies in those aged 15 to 34 years. 5 The risk of developing unprovoked seizures after head trauma is directly related to the severity of the injury. 6 Other risk factors include penetrating injuries, biparietal or multiple brain contusions, intracranial hemorrhage, frontal or temporal location of the lesion, age >65 years at the time of injury, and a past history of depression. 6 Remarkably, there is ample variability in brain responses to similar injuries: Most patients will not develop seizures, some will experience one or few seizures, and others will have frequent seizures and drug-resistant epilepsy. 7 Such a wide range of variability has been attributed to “constitutional” or genetic factors influencing the cerebral response to injury. 7
Early studies attempted to characterize genetic contributions to post-traumatic epilepsy by including a family history of seizures or epilepsy in risk factor analyses, yielding conflicting results. 8 These studies were hampered by methodological limitations, such as selection bias, small sample size, short follow-up after head trauma, ambiguous definitions of head trauma or epilepsy, and reliance on patient reporting to collect information about family history. 8 These shortcomings were addressed by Christensen et al, 9 who conducted a population-based study of >1.6 million people born in Denmark (1977-2002) to assess the risk of epilepsy up to ≥10 years after traumatic brain injury. They found that the risk of epilepsy following any type of head trauma was 2.3-fold higher among individuals who had a family history of epilepsy (defined as having a parent or sibling who had been in outpatient care or hospitalized with a diagnosis of epilepsy, ascertained using International Classification of Diseases codes) compared to those without such a family history (adjusted relative risk [95% CI]: 5.73 [4.58-7.16] vs 2.47 [2.31-2.65]). Importantly, a family history of epilepsy conferred a substantially increased risk of epilepsy even following mild head trauma (adjusted relative risk [95% CI]: 5.75 [4.56-7.27]), albeit lower than for severe head trauma (10.09 [4.20-24.26]).
Variation in a number of genes has been suggested to raise the risk of post-traumatic epilepsy, including IL-1β (encoding the pro-inflammatory cytokine interleukin-1β), 10 MTHFR (encoding the enzyme methylenetetrahydrofolate reductase), 11 SLC1A3 (encoding the neuronal excitatory amino acid transporter 1), 12 GAD1 (encoding the glutamic acid decarboxylase-67 responsible for γ-aminobutyric acid synthesis from glutamate), 13 and genes implicated in adenosine homeostasis (ADK, NT5E, and A1AR; Table 1). 14,15 However, these findings were obtained from small studies which examined selected single-nucleotide polymorphisms (SNPs) within 1 to 3 genes, using a hypothesis-driven candidate gene approach. Critically, no gene association study in post-traumatic epilepsy has included a replication cohort. Thus, these results should be viewed as preliminary, requiring validation in larger patient cohorts. The epilepsy genetics literature comprises many examples of genes claimed to be associated with epilepsy, which do not withstand replication. 16 This might also be the case for the suggested increased risk of post-traumatic epilepsy conferred by the APOE ε4 allele, 17 which was not replicated in 4 other studies (Table 1). 18 -21
Gene association studies in post-traumatic epilepsy.
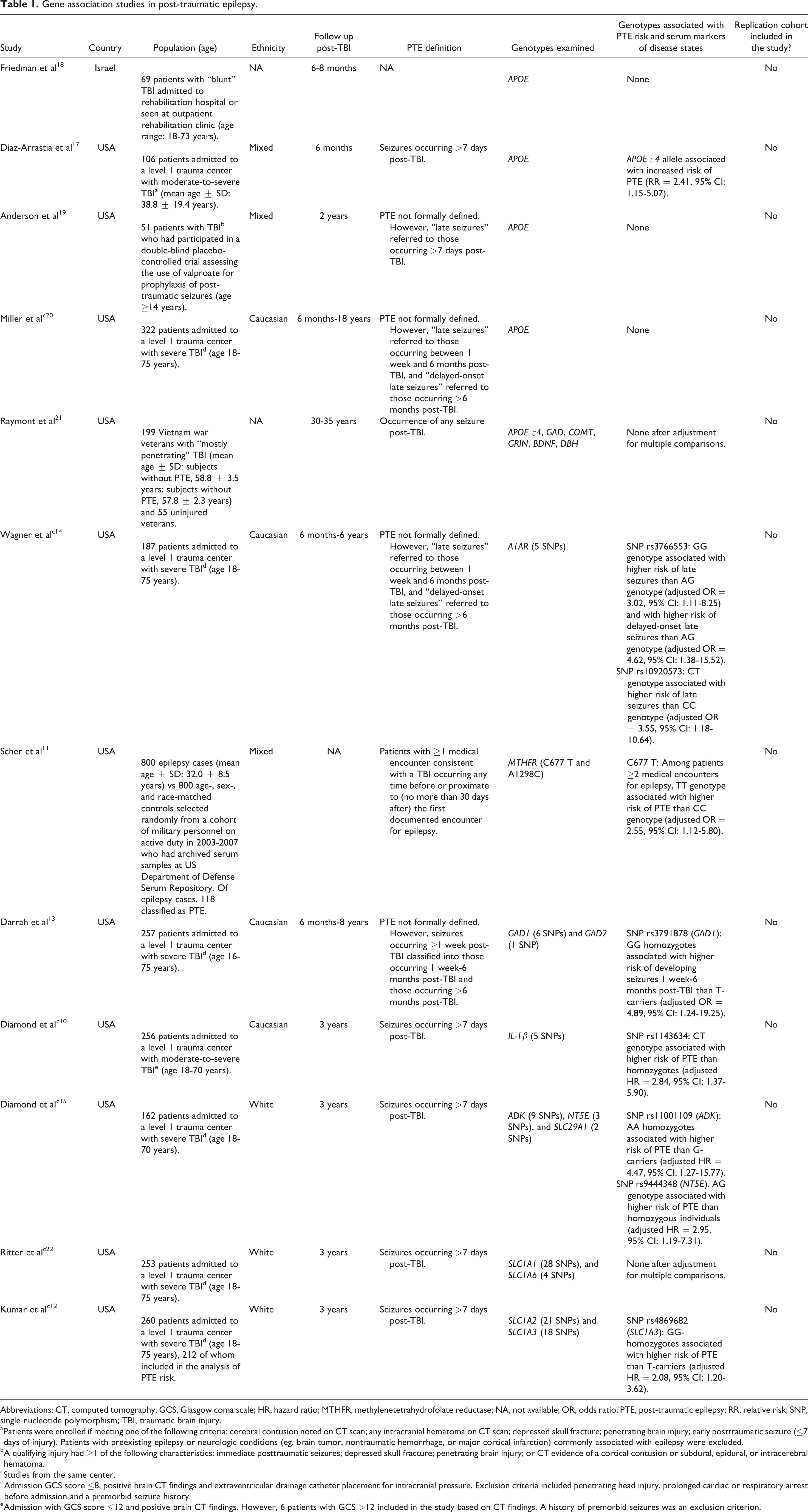
Abbreviations: CT, computed tomography; GCS, Glasgow coma scale; HR, hazard ratio; MTHFR, methylenetetrahydrofolate reductase; NA, not available; OR, odds ratio; PTE, post-traumatic epilepsy; RR, relative risk; SNP, single nucleotide polymorphism; TBI, traumatic brain injury.
a Patients were enrolled if meeting one of the following criteria: cerebral contusion noted on CT scan; any intracranial hematoma on CT scan; depressed skull fracture; penetrating brain injury; early posttraumatic seizure (≤7 days of injury). Patients with preexisting epilepsy or neurologic conditions (eg, brain tumor, nontraumatic hemorrhage, or major cortical infarction) commonly associated with epilepsy were excluded.
b A qualifying injury had ≥1 of the following characteristics: immediate posttraumatic seizures; depressed skull fracture; penetrating brain injury; or CT evidence of a cortical contusion or subdural, epidural, or intracerebral hematoma.
c Studies from the same center.
d Admission GCS score ≤8, positive brain CT findings and extraventricular drainage catheter placement for intracranial pressure. Exclusion criteria included penetrating head injury, prolonged cardiac or respiratory arrest before admission and a premorbid seizure history.
e Admission with GCS score ≤12 and positive brain CT findings. However, 6 patients with GCS >12 included in the study based on CT findings. A history of premorbid seizures was an exclusion criterion.
Poststroke Epilepsy
Stroke is the leading cause of acquired epilepsy, accounting for almost one-third of acquired epilepsies. 5 Its relevance is even greater in the elderly (age >65 years), where it accounts for 2 of 3 new cases of acquired epilepsy. 5 Stroke characteristics, namely stroke severity, cerebral hemorrhage, and cortical involvement, are the key risk factors for poststroke epilepsy. 23 Other suggested risk factors include early seizures, occurring within 7 days of stroke and younger age at stroke. 23
A family history of epilepsy also increases the risk of poststroke epilepsy, 24,25 although to a lesser degree than stroke characteristics. In a recent Swedish, register-based, cohort study of 86 550 adults with stroke, the survival-adjusted risk of late poststroke seizures (ie, seizures occurring more than 7 days after stroke, corresponding in most cases to poststroke epilepsy) was higher among patients who had ≥1 first degree relative with epilepsy compared to those without: 6.8% (95% CI: 6.2-7.4) versus 5.9% (95% CI: 5.7-6.1) at 2 years and 9.5% (95% CI: 8.7-10.3) versus 8.2% (95% CI: 8.0-8.4) at 5 years. 25 In an analysis adjusting for age, sex, stroke type and number of relatives, the hazard ratio for late poststroke seizures in patients with ≥1 first degree-relative with epilepsy was 1.18 (95% CI: 1.09-1.28). These findings were retained after adjusting for stroke severity and other potential confounders. In particular, the authors excluded that the increased risk of late poststroke seizures conferred by a family history of epilepsy was attributable to a familial predisposition to stroke.
Four studies have explored susceptibility genes for poststroke epilepsy (Table 2). 26 -29 Variation in 3 genes was found to increase the risk of poststroke epilepsy, that is, ALDH2 (encoding the enzyme mitochondrial aldehyde dehydrogenase 2), CD40 (encoding the costimulatory receptor CD40), and TRPM6 (encoding the transient receptor potential melastatin type 6 magnesium-permeable channel). 27 -29 Each genetic variation was associated with altered serum concentrations of a specific molecule, which were postulated to mediate its effects (Table 2). However, these studies had several major limitations potentially compromising the validity of their findings, including small sample sizes; lack of characterization of the stroke subtype(s) in the study populations; failure to detail the follow-up after stroke and if there were any differences in length of follow-up between patients who developed unprovoked seizures and those who did not; ambiguous definitions of poststroke epilepsy; and failure to adjust analyses for potential confounders. Furthermore, each of these studies examined a specific SNP only (typically without adequately justifying the selection) and did not include a replication cohort. Thus, replication of their results is sorely needed.
Gene Association Studies in Poststroke Epilepsy.
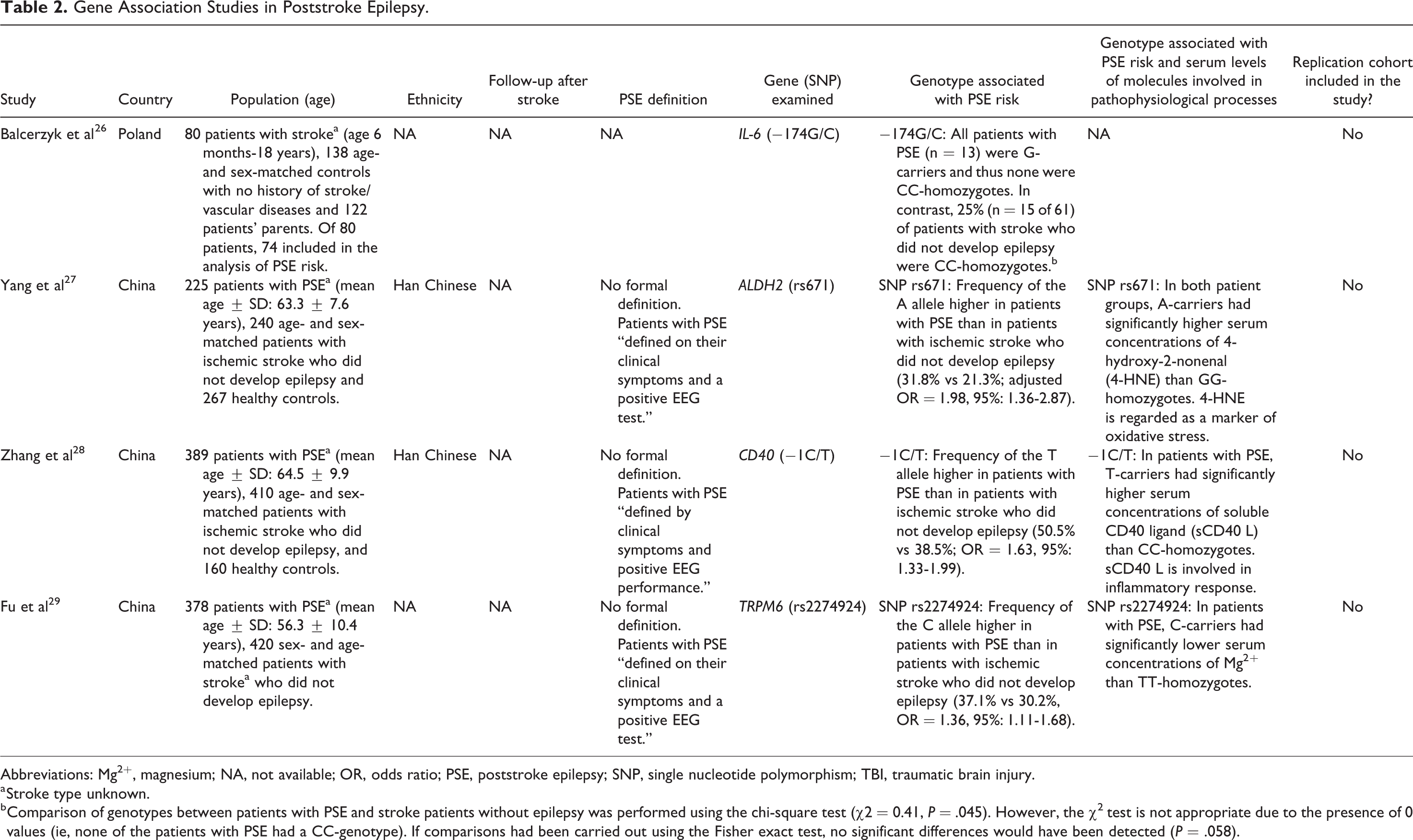
Abbreviations: Mg2+, magnesium; NA, not available; OR, odds ratio; PSE, poststroke epilepsy; SNP, single nucleotide polymorphism; TBI, traumatic brain injury.
a Stroke type unknown.
b Comparison of genotypes between patients with PSE and stroke patients without epilepsy was performed using the chi-square test (χ2 = 0.41, P = .045). However, the χ2 test is not appropriate due to the presence of 0 values (ie, none of the patients with PSE had a CC-genotype). If comparisons had been carried out using the Fisher exact test, no significant differences would have been detected (P = .058).
Temporal Lobe Epilepsy With Hippocampal Sclerosis
Hippocampal sclerosis is the most common histopathological substrate in drug-resistant temporal lobe epilepsy, 30 and its resection or destruction often results in seizure freedom. 31 The pathogenesis of hippocampal sclerosis has long been debated and remains incompletely understood. Prolonged febrile seizures, including febrile status epilepticus, have been associated with acute hippocampal injury and subsequent development of hippocampal sclerosis and temporal lobe epilepsy. 32 In some children with prolonged febrile seizures, however, there are preexisting hippocampal abnormalities which may predispose to hippocampal damage. 33 The prospective FEBSTAT (Consequences of Prolonged Febrile Seizures in Childhood) study suggests that both mechanisms may be operative. 32,33
Genetic factors are also implicated. Children with their first febrile seizure had a 3-fold higher risk of presenting with febrile status epilepticus than a simple febrile seizure if they had a first-degree family history of febrile seizures. 34 An increased prevalence of a family history of febrile and afebrile seizures has been observed in patients with temporal lobe epilepsy and hippocampal sclerosis. 35 Furthermore, a clinically heterogeneous form of familial mesial temporal lobe epilepsy, often characterized by antecedent febrile seizures, hippocampal sclerosis, and drug resistance, has been described. 36,37 In these families, most individuals with drug-resistant epilepsy and hippocampal sclerosis have favorable seizure outcomes after epilepsy surgery, similarly to sporadic patients. 38
A number of early studies investigated the role of common susceptibility variants in temporal lobe epilepsy with hippocampal sclerosis, suggesting an association with SNPs in the IL-1β and PRNP genes. 39,40 However, these findings failed to be replicated in larger cohorts. 16 A subsequent 2-stage genome-wide association study (GWAS) including 1977 cases with mesial temporal lobe epilepsy and hippocampal sclerosis and 11 143 controls of European Ancestry provided suggestive evidence for common variation in the SCN1A gene (encoding the α-subunit of the type 1 voltage-gated sodium channel) to increase susceptibility to mesial temporal lobe epilepsy with hippocampal sclerosis and febrile seizures. 41 Recently, the International League Against Epilepsy (ILAE) Consortium on Complex Epilepsies published its second meta-analysis of GWASs involving 15 212 cases with epilepsy and 29 677 controls, which identified 2 novel loci for focal epilepsy with hippocampal sclerosis: 3q25.31, implicating C3orf33, SLC33A1 or KCNAB1, and 6q22.31, implicating the gap junction gene GJA1. 42
A recent study examined the role of rare and de novo variants in mesial temporal lobe epilepsy with hippocampal sclerosis by applying whole-exome sequencing (WES) to 47 Han Chinese patients, including 23 patient–parent trios. 43 Compared to WES data from 692 Han Chinese controls, significant enrichment of rare variants was observed in SEC24B, a gene involved in vesicle trafficking and development. Gene-set association analysis showed variant enrichment in the fragile X mental retardation protein–related group of genes. Trio-based analysis detected 21 de novo variants, many of which have been implicated in neuropsychiatric disorders. Overall, these results should be interpreted with caution due to the study’s small sample size, and their replication in larger cohorts is awaited.
Other Acquired Epilepsies
Genetic influences have been investigated in other types of acquired epilepsy. Three independent groups recently described the association of anti-leucine-rich glioma-inactivated 1 (anti-LGI1) encephalitis with the human leukocyte allele (HLA)-II alleles DRB1*07:01 and DQB1*02:02. 44 -46 Two groups detected this association by applying selective HLA genotyping, 44,45 whereas the third group performed a GWAS. 46 Remarkably, these findings were obtained despite the inclusion of only a small number of patients with anti-LGI1 encephalitis in each study (19 Dutch, 11 Korean, and 54 German patients, respectively). 44 -46 Each study also found other HLA alleles, that is, DRB4, DQA1*02:01, B*44:03, increased susceptibility to anti-LGI1 encephalitis, but replication is warranted.
Most recently, a small study explored genetic contributions to nodding syndrome, a rare disorder characterized by nodding head, epileptic seizures, stunted growth, and neurological deterioration, affecting children aged 5 to 16 years in Tanzania, Uganda, and Republic of South Sudan. 47 The disorder has been linked to an infection by the parasitic worm Onchocerca volvulus. 47 Benedek et al 48 analyzed 7 HLA loci in 48 cases with nodding syndrome versus 51 controls from South Sudan and suggested that 5 HLA alleles, that is, B*42:01, C*17:01, DRB1*03:02, DQB1*04:02, and DQA1*04:01, might confer protection from the disorder. Due to the limited sample size, these findings should be viewed as preliminary, requiring replication in larger cohorts.
Conclusions
Over the past decade or so, there has been progress in elucidating genetic contributions to acquired epilepsy. Two well-designed epidemiological studies have quantified the increased risk of developing unprovoked seizures after a traumatic brain injury 9 or stroke 25 in the setting of a family history of epilepsy. Many genetic variants have been suggested to increase susceptibility to certain forms of acquired epilepsy. However, most of these variants were identified in studies which were hampered by many limitations, including small sample size, unclear definitions, and the investigation of only selected genes. Therefore, replication of these findings is required.
Considering that disease susceptibility alleles have small effect sizes, large collaborative efforts are needed to assemble adequately sized cohorts to investigate genetic variants raising the risk of acquired epilepsy. Large collaborative frameworks are already a reality in epilepsy genetics research and are leading important discoveries. 42,49 The application of novel approaches, such as polygenic risk scores, will also advance the field. 50 Undoubtedly, genetic influences in acquired epilepsy are only starting to be unraveled. With the expanding opportunities to investigate disease susceptibility variants, further progress is expected in the upcoming years. This promises to unlock insights into the underlying mechanisms and enable development of targeting medicine to prevent epilepsy following acquired brain injury.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.