
Abstract
Gene therapy has recently advanced to the level of standard of care for several diseases. However, its application to neurological disorders is still in the experimental phase. In this review, we discuss recent advancements in the field that provide optimism on the possibility to have first-in-human studies for gene therapy of some forms of epilepsy in the not so distant future.
Keywords
Introduction
As is well known, current therapies for epilepsy are largely unsatisfactory. 1 In spite of the many available antiepileptic drugs and of other therapeutic approaches (surgery, brain stimulation, ketogenic diet, etc), about one-third of the patients do not get control of their seizures. We do not have any treatment able to prevent epilepsy development in at-risk individuals. We do not have adequate control of epilepsy comorbidities that heavily affect the quality of life of patients. Many devastating forms of epilepsy are resistant to any treatment. And the list could continue.
Within this scenario, the search for new, alternative therapeutic approaches is always a priority, and gene therapy is often a consideration. In principle, the idea is simple: Use some kind of vector to transfer the DNA encoding some “therapeutic” protein(s) into the diseased cells, in order to permanently heal them. There are different types of DNA that one could desire to transfer, reflecting different therapeutic strategies. The most obvious is the healthy variant of a defective gene, which could be an option in some genetic forms of epilepsy. But it would also be possible to attempt healing the defective gene using gene editing technologies, including clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9-mediated genetic modification, and gene activation or inhibition (CRISPRa or CRISPRi). Another option could be to transfer genes that can modify cell (or circuit) function and control hyperexcitability, such as channels, neurotransmitters, or receptors. Finally, one could transfer genes encoding proteins that render the cell sensitive to specific drugs (chemogenetics) or to light stimulation (optogenetics).
In sum, there are multiple diverse options and strategies on stage. But are these doable? Can they be applied to all forms of epilepsy? Which forms of epilepsy may represent low hanging fruit for starting a program of clinical translation? Answering the first question means having at hand systems of gene transfer (vectors) that are safe, allow transfer of a sufficiently large DNA cargo, and ensure robust, lasting, and regulated expression of the therapeutic gene(s) in a specific target cell. The other questions underlie other key problems, for example, the fact that, for focal epilepsies, it may be sufficient to inject the vector and express the therapeutic gene(s) in a relatively restricted brain area, whereas in generalized epilepsies there could be a need to obtain widespread expression in the whole brain.
Below, we briefly summarize the state of the art of vector development for gene therapy and the results of preclinical studies in epilepsy models. We then describe some recent advances that may be implemented in epileptology, moving the field closer to a much-awaited clinical application.
Viral Vectors for the Central Nervous System
There are 2 main classes of gene delivery tools, nonviral and viral vectors, each endowed with specific advantages and disadvantages. Compared with viral vectors, nonviral vectors tend to have lower immunogenicity, due to the absence of preexisting immunity, larger payload capacity, and easier production techniques. 2,3 However, their major drawback is the low transduction efficiency.
In contrast, viral vectors exploit the viruses’ highly evolved strategies for efficient transfer of foreign DNA into eukaryotic cells. Among different viruses engineered and tested for gene therapy, the most promising candidates for central nervous system (CNS) applications seem to be adeno-associated viruses, lentiviruses and herpes viruses (Table 1). Adeno-associated viruses (AAV) are small single-stranded DNA viruses. 4,5 In spite of their limited cargo capacity (4.5 kb), AAV vectors are the most commonly used in clinical trials for CNS gene therapy 6 because they exhibit low immunogenicity, no pathogenicity, and long-lasting transgene expression in both dividing and nondividing cells. 7 Several AAV serotypes have been identified and developed, based on capsid variants that confer different tropisms, antigenic profiles, 8 and transduction efficiency. 9,10 For example, the AAV1, AAV2, AAV5, and AAV8 serotypes display a marked neuronal tropism, 11 -13 whereas the AAV9 serotype can cross the blood–brain barrier (BBB) after peripheral administration. 14 A tropism shift from neurons to glia is observed in the mature brain, 15 indicating that brain development should be considered for therapeutic applications. All these features can be modulated and improved by combining 2 or more different serotypes, 8 by mutation of capsid tyrosine residues, 16 or by fusing peptides to capsid proteins. 6 One major problem with AAV vectors is the inactivation by neutralizing antibodies. However, chemical compounds 8 or association with exosomes 17 have been tested to shield the capsid from neutralizing antibodies.
Viral Vectors.
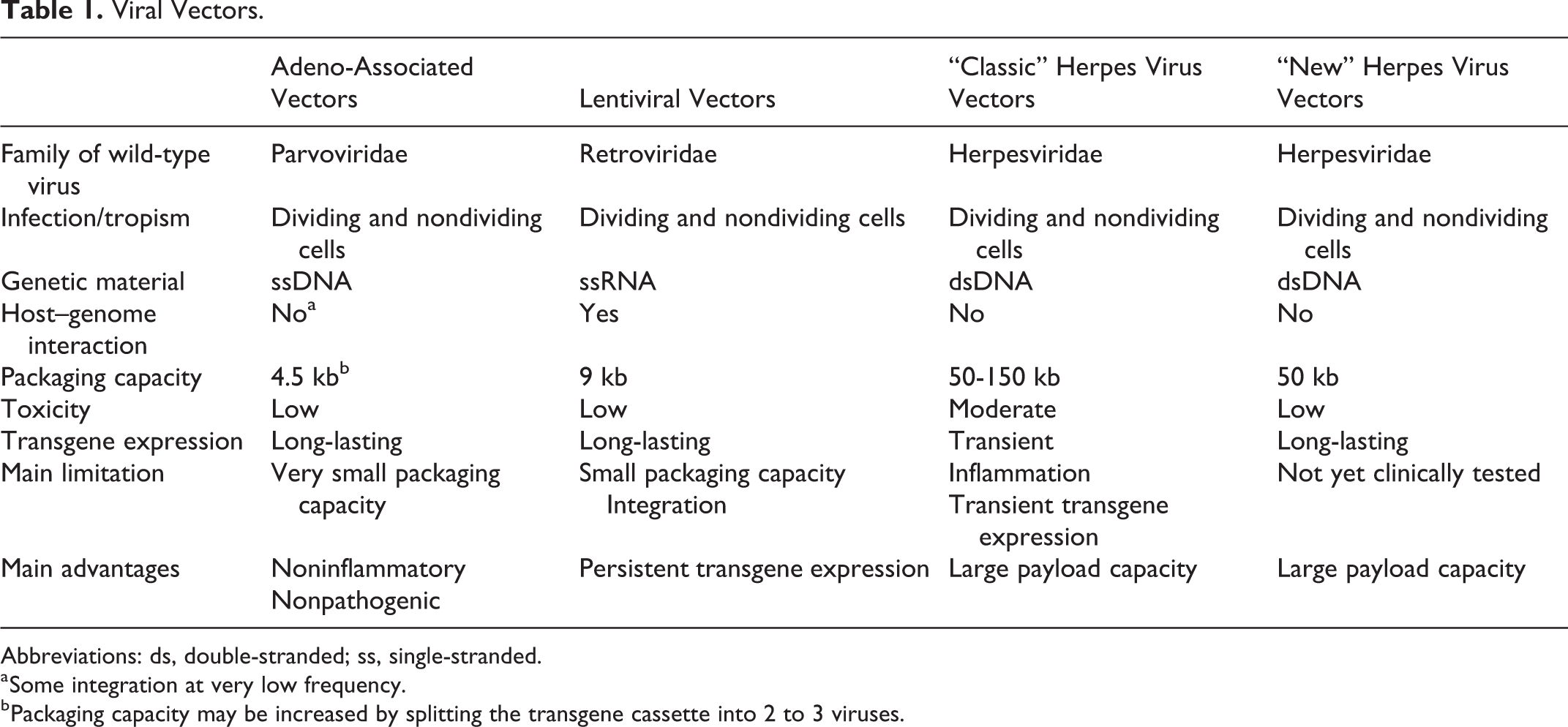
Abbreviations: ds, double-stranded; ss, single-stranded.
a Some integration at very low frequency.
b Packaging capacity may be increased by splitting the transgene cassette into 2 to 3 viruses.
Lentiviruses (LV) are integrating single-stranded RNA viruses, capable of transducing nondividing and dividing cells. 18 Most LV vectors derive from human immunodeficiency virus and have a transgene payload capacity of about 9 kb. 19 Pseudotyping their envelope with glycoproteins derived from herpes simplex virus (HSV), rabies, or vesicular stomatitis virus allows one to modify, improve, and refine cellular tropism and transduction efficiency. 20 -24 Insertional mutagenesis is a potential risk. Nonintegrating LV vectors have been developed by introducing mutations in the integrase gene, such that the viral genome persists in the host cell mostly (even if not exclusively) in an episomal form. 25
The HSV vectors are double-stranded DNA viruses that can be divided into 3 main categories: replication-competent (employed in cancer), replication-defective, and amplicon vectors, carrying a DNA plasmid instead of the viral genome. 26 Both replication-defective and amplicon vectors display natural neuronal tropism and high payload capacity, up to 50 and 150 kb, 27 respectively, which allows the insertion of large transgenes and regulation systems. These features, together with a high transduction efficiency, the ability of both anterograde and retrograde transport, and the episomal nonintegrating genome, make HSV vectors an attractive tool for CNS disorders. 26 -28 Their downsides are the residual toxicity and a relatively transient transgene expression. 27 In addition, preparations of amplicon vectors remain contaminated by a small percentage of helper virus. 28 However, new generation vectors seem to overcome these problems (Table 1 and see below).
Gene Therapy Approaches in Animal Models of Epilepsy
All work on gene therapy for epilepsy thus far has been performed in animal models and by focal administration of vectors. In most cases, the focus has been on post-status epilepticus (SE) models. One study explored the antiepileptogenic effect of HSV vector-mediated delivery of neurotrophic factors such as fibroblast growth factor 2 and brain-derived neurotrophic factor in the pilocarpine model. 29 Injection of this vector into the hippocampus during the latent period (3 days after SE) attenuated seizure-induced damage, favored a more physiological neurogenesis, and highly reduced the occurrence of spontaneous recurrent seizures (SRSs).
All other studies focused on the chronic period, when animals were experiencing SRSs, and used seizure frequency as the primary outcome measure. This approach has greater translational potential, as it may be offered to individuals with drug-resistant focal seizures that cannot be treated surgically. Overall, the aim of all these studies was to increase inhibition in the epileptogenic area, but strategies were diverse. For example, some used AAV vectors to downregulate excitatory receptor function (by transferring antisense NR1, an essential subunit of the NMDA receptors) or to upregulate inhibitory receptor function (by transferring the α-1 subunit of the GABAA receptor). 30,31 Others used LV vectors to overexpress potassium channels or halorhodopsin for inhibitory optogenetic stimulation. 32 Or, using AAV vectors, one group expressed a modified muscarinic receptor (hM4Di) to obtain seizure suppression by systemic administration of the hM4Di selective, normally inactive agonist clozapine-N-oxide. 33 Yet another strategy was to express a genetically modified glutamate-gated Cl− channel. 34
All these strategies proved effective. However, a common challenge is the ability to express the transgene in a specific cell population, because inhibiting inhibitory neurons would favor the occurrence of seizures. 30 To avoid this problem, therefore, these authors drove expression of their transgenes by promoters specifically active in excitatory neurons, in an attempt to bias expression toward (and thereby preferentially inhibit) excitatory neurons. As discussed below, this is a reasonable but imperfect solution.
One alternative strategy is the expression of a soluble inhibitory factor that can be secreted by the transduced cells: In this case, seizure control may be achieved without targeting specific cells, provided that the receptors for that factor are found in the injected area. 35 Several studies have demonstrated that overexpression in the hippocampus of inhibitory neuropeptides (neuropeptide Y [NPY], galanin, or somatostatin) exerts antiseizure effects in epilepsy models (data on NPY and galanin reviewed in Simonato, 35 Simonato et al, 36 and Kullmann et al 37 ; for somatostatin, see Natarajan et al 38 ).
Among these neuropeptides, NPY seems the most promising translationally because it is the most effective in suppressing seizure-like activity in slices from the human epileptic hippocampus. 39 However, a complication (and an opportunity) for NPY is that its effects are mediated by multiple receptors, some proepileptic (the Y1 subtype), others (Y2 and Y5) antiepileptic. 40 A combined administration of an AAV vector expressing NPY with one expressing the Y2 or one expressing the Y5 receptor produced much stronger reductions in seizure frequency than NPY alone. 41,42
Looking Forward
Altogether, the results of preclinical studies in epilepsy models suggest optimism as to the possibility of translation in humans. This optimism is sustained by recent advancements in clinical experimentation for other CNS diseases, in vector design, and in targeting and regulation strategies.
Advancements in clinical experimentation for CNS diseases
The main obstacles on the way to human translation are the complexity and heterogeneity of the target tissue, the presence of the BBB, and the safety of viral vectors. However, successful reports from experimentation for other CNS diseases are helping to concretely chart out a roadmap toward the first-in-man gene therapy for intractable epilepsy. 43 -45
The direct intraparenchymal infusion of viral vectors has been successfully explored in a number of clinical studies for neurological disorders. 36 One of the most promising envisaged the bilateral injection in the subthalamic nucleus of patients affected by medically refractory Parkinson disease with a mix of recombinant AAVs encoding GABA synthesizing enzymes (GAD65 or GAD67). Upon assessment of safety and tolerability, 46 this study became the first double-blinded and randomized trial of gene therapy for the CNS, 47 showing beneficial effects on motor function that persisted up to 12 months. 48
A more recent study explored the use of an LV gene therapy vector for the simultaneous delivery into the striatum of 3 key enzymes for dopamine biosynthesis, providing a local and sustained novel source of dopamine from nondopaminergic transduced cells. A first trial positively verified the safety profile of this treatment, 49 which was subsequently confirmed in an 8-year follow-up along with a moderate improvement of motor function. 50
Another major advancement was the discovery of the ability of the AAV9 serotype to cross the BBB, which makes it potentially usable to treat genetic neurological diseases by transferring the healthy allele to the brain in a widespread manner. In a phase 1 clinical study, 51 a group of patients affected by spinal muscular atrophy type-1 (SMA1), a monogenic disorder caused by mutation of the survival motor neuron-1 (SMN1) gene, has been successfully treated by a single, systemic dose of a recombinant AAV9 vector carrying the SMN1 gene. In principle, this approach may be used for some monogenetic forms of epilepsy, such as Dravet syndrome (DS). Similar to SMA1, DS is generally caused by heterozygous mutations of the gene encoding the voltage-gated sodium channel α1 subunit (SCN1A). Unfortunately, however, neither the AAV nor the LV vectors can accommodate the entire SCN1A expression cassette in a single vector. To overcome this hurdle, the AAV packaging capacity may be increased by harnessing the virus natural propensity to generate head-to-tail DNA concatamers in the infected cells. 52,53 This feature has been exploited to split and package large transgene cassettes in 2 54 -56 or 3 57 separate AAV viral particles. The full-length cassette can then be recovered in cells that are concomitantly infected by the whole set of vectors; however, this approach significantly reduces the efficiency of gene transfer. 55 Efforts are ongoing to mitigate this problem. 56,57
Advances in vector design
Vectors for CNS gene therapy should be highly refined, in order to ensure delivery to specific cell types, efficacy of transgene expression, capacity to host large and/or multiple inserts, safety, lasting transgene expression, and mechanisms to regulate expression. As described above, AAV and LV vectors do not combine all these features, while HSV vectors have been relatively overlooked so far because of concerns about cytotoxicity, immunogenicity, and difficulty in achieving persistent expression in the CNS. However, we have recently developed a new generation of HSV vectors that overcome these problems (Table 1). These are highly replication-defective vectors, devoid of all viral immediately early genes, in which viral gene expression is virtually absent. 58 We found that inserting an expression cassette in a specific locus of the genome (the ICP4 locus) permits robust and long-term reporter gene expression in a diversity of neurons following stereotactic injection in the brain. 59 Virus infection did not cause any neurotoxicity or inflammatory infiltrates. Therefore, these are high-capacity vectors capable of safe, long-term transgene expression in the brain, opening up the possibility for therapeutic intervention into CNS diseases that require transfer of large amounts of DNA.
Advances in cell targeting and in regulation strategies
Other key advances were recently made for achieving a robust expression of the transgenes in a cell-specific manner, avoiding the risk of off-target effects. As mentioned above, the “classic” approach is to drive transgene expression through promoters that are active only in the desired cell type. 60 For example, candidates to restrict gene expression in inhibitory neurons are the GAD65 or GAD67 promoters that code for the enzyme that catalyzes the transformation of glutamate into GABA. However, this procedure does not completely ensure selectivity of expression and, in addition, the size of many full-length promoters is too large for most viral vectors. An alternative and more efficient strategy was recently proposed, based on the encoding of microRNA target motifs downstream of the transgene. The introduction of multiple target motifs for microRNAs expressed in off-target cells silenced transgene expression in these cells, thereby achieving highly specific expression in the desired cell type. 61
Another important advance involves the regulation of gene expression. Mechanisms of autoregulation are important requirements for clinical translation because they reduce the risk of negative effects on physiological brain circuitries. Lieb et al 34 cloned into an LV vector an optimized sequence encoding a glutamate-gated chloride channel with an EC50 for glutamate of about 10 μM, that is, high concentrations that would be reached at extrasynaptic levels only during seizures. When injected in the rat neocortex, this vector led to a potent attenuation of evoked and spontaneous seizures, in the absence of alterations in normal brain function.
Conclusions
Although gene therapy is becoming an established approach for an increasing number of diseases, its application to CNS disorders still poses formidable challenges that are not yet completely overcome. Although it will be essential to sort out all possible problems before clinical testing, the good news is that the field is progressing rapidly and it seems plausible that the time for a first-in-man gene therapy for epilepsy is not too far anymore.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work by the authors described in this review was supported by the FP7-HEALTH project 602102 (EPITARGET).