
Abstract
Introduction:
A dramatic increase in the number of clinical trials involving gene-modified cell therapy and gene therapy is taking place. The field is on the verge of a boom, and the regulatory environment is evolving to accommodate the growth.
Discussion:
This commentary summarizes the current state of the field, including an overview of the growth. The United States (US) regulatory structure for gene therapy will be summarized, and the evolution of the oversight structure will be explained.
Conclusion:
The gene therapy field has recently produced its first FDA-approved therapeutics and has a pipeline of other investigational products in the final stages of clinical trials before they can be evaluated by the FDA as safe and effective therapeutics. As research continues to evolve, so must the oversight structure. Biosafety professionals and IBCs have always played key roles in contributing to the safe, evidence-based advancement of gene therapy research. With the recent regulatory changes and current surge in gene therapy research, the importance of those roles has increased dramatically.
Keywords
Introduction
For decades, gene therapy was an elusive goal relegated to the world of science fiction.
Concerns over the nascent field of genetic engineering and the risks associated with human gene transfer led to the Asilomar conference in 1975, where leading researchers debated the risks and proposed a framework for oversight. This framework was later adopted as NIH Guidelines. The NIH also created the Recombinant DNA Advisory Committee (RAC) to advise the director of the NIH on topics pertaining to emerging biotechnology.
The field experienced highly publicized setbacks in the late 1990s and early 2000s, including the French SCID trial and the death of Jesse Gelsinger, which made some question the feasibility of human gene transfer. Since the turn of the century, however, much progress has been made in understanding the risks associated with gene transfer technology and developing safety features. In recent years, FDA has also established various guidance documents for gene therapy research.
Several additional factors point to an environment where the gene therapy field is ready to boom in the clinical setting: advancements in biotechnology, a greater emphasis on translational medicine, investments from the pharmaceutical sector, and an evolving regulatory environment. An explosion in gene therapy research appears to be on its way. This commentary details how the US regulatory environment is adapting to support this upsurge.
Discussion
The Current State of the Gene Therapy Field in US
To date, over 2900 gene therapy studies have been initiated worldwide, according to the Journal of Gene Medicine. 1 Searching clinicaltrials.gov for the keywords gene therapy results in more than 3800 studies with over 1000 of these studies currently recruiting or enrolling research subjects. 2
As the field focused on reassessing safety, the number of investigational new drug (IND) applications for gene therapy products submitted to the FDA reached a period of plateau from 1995 to 2010 (Figure 1 3 ). However, this number has been steadily climbing since 2011. The year 2018 marked an all-time high number of IND applications (206), almost doubling the previous all-time high achieved in 2017 (106). The surge in research has also led to approvals; the FDA issued its first approval of a product containing recombinant DNA in 2015, issued 3 gene therapy approvals in 2017, and recently issued two more in May of 2019. With 291 gene therapy studies currently in phase III, several more gene therapy products are expected to be considered for approval in the coming years.
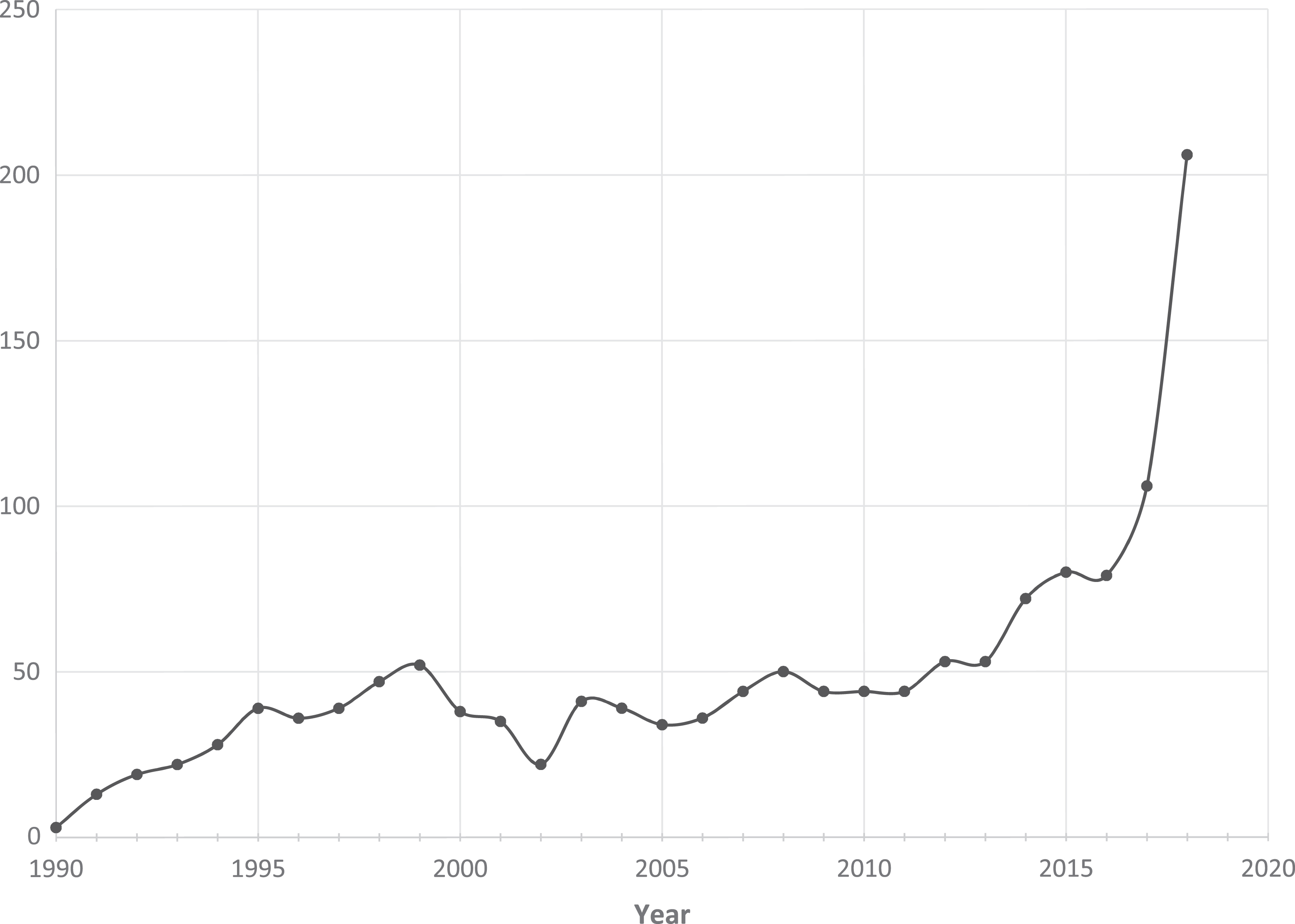
Gene therapy IND applications submitted per year. Data adapted with permission from Peter Marks, Director, FDA Center for Biologics Evaluation and Research (CBER). 3
US Regulatory Oversight Structure for Gene Therapies
A summary of the structure for regulatory oversight of investigational products for human gene transfer is provided in Figure 2. Initial research and development of gene therapy products typically begins in academic or industry laboratories focused on demonstrating a proof of concept. Preclinical safety data are first generated in a laboratory compliant with Good Laboratory Practice (GLP) regulations. When researchers are ready to begin clinical testing in human subjects, an IND application must be submitted to the FDA for authorization to administer an investigational drug to humans. Such authorization must be secured prior to the interstate shipment and administration of any new unapproved drug.
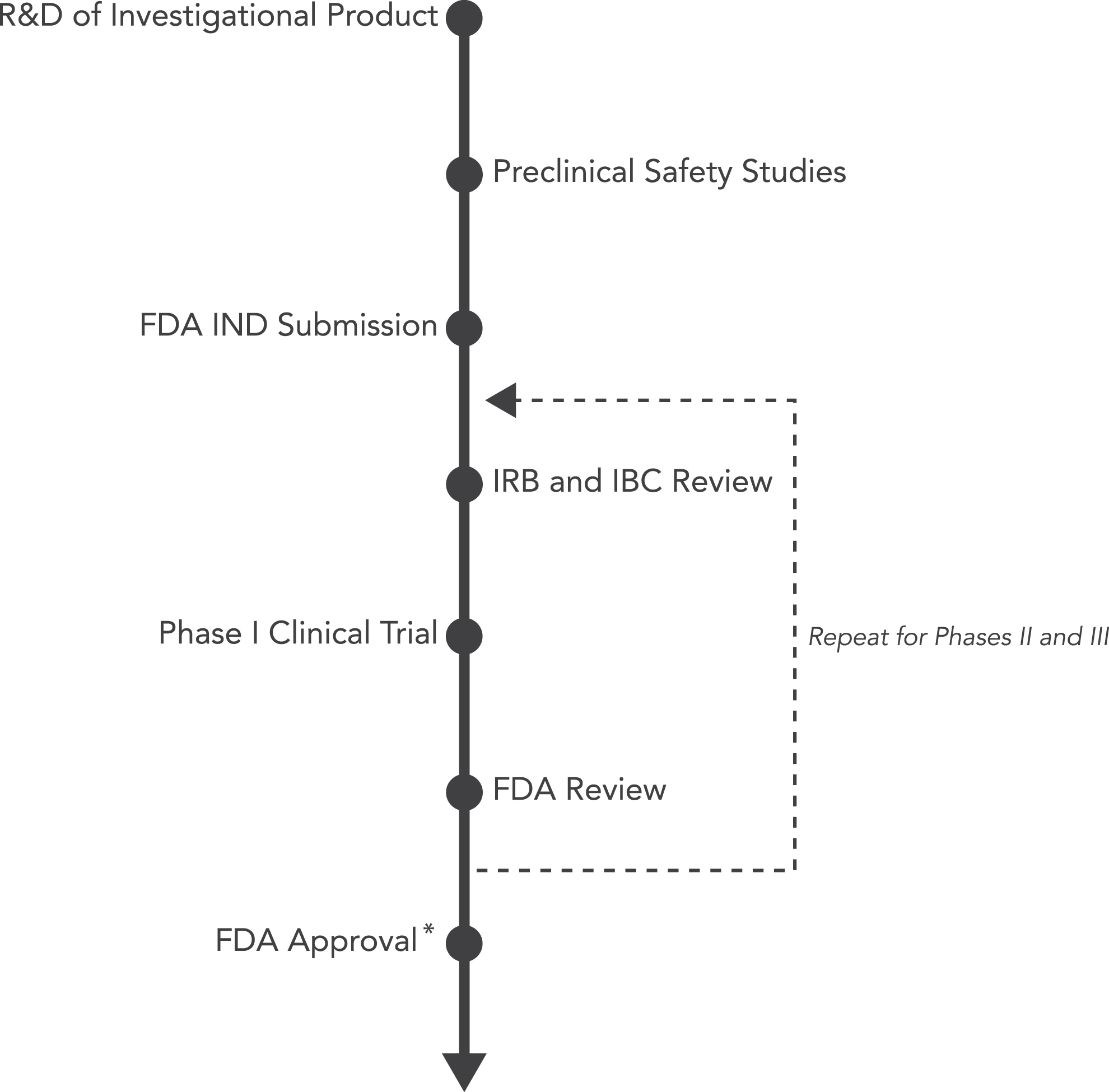
The regulatory approval process for investigational products for human gene transfer studies. *IBC approval is not required for on-label use of FDA-approved gene therapy products as such use does not constitute research.
After receiving an IND number from the FDA, the research sponsor applies for institutional review board (IRB) approval. The IRB will review the proposed clinical trial to ensure it conforms with regulations for human subject protection. If NIH funding is associated with the research and development of the investigational product, the clinical trial, or the research site(s) conducting the study, institutional biosafety committee (IBC) approval is required at each research site to ensure compliance with NIH guidelines.
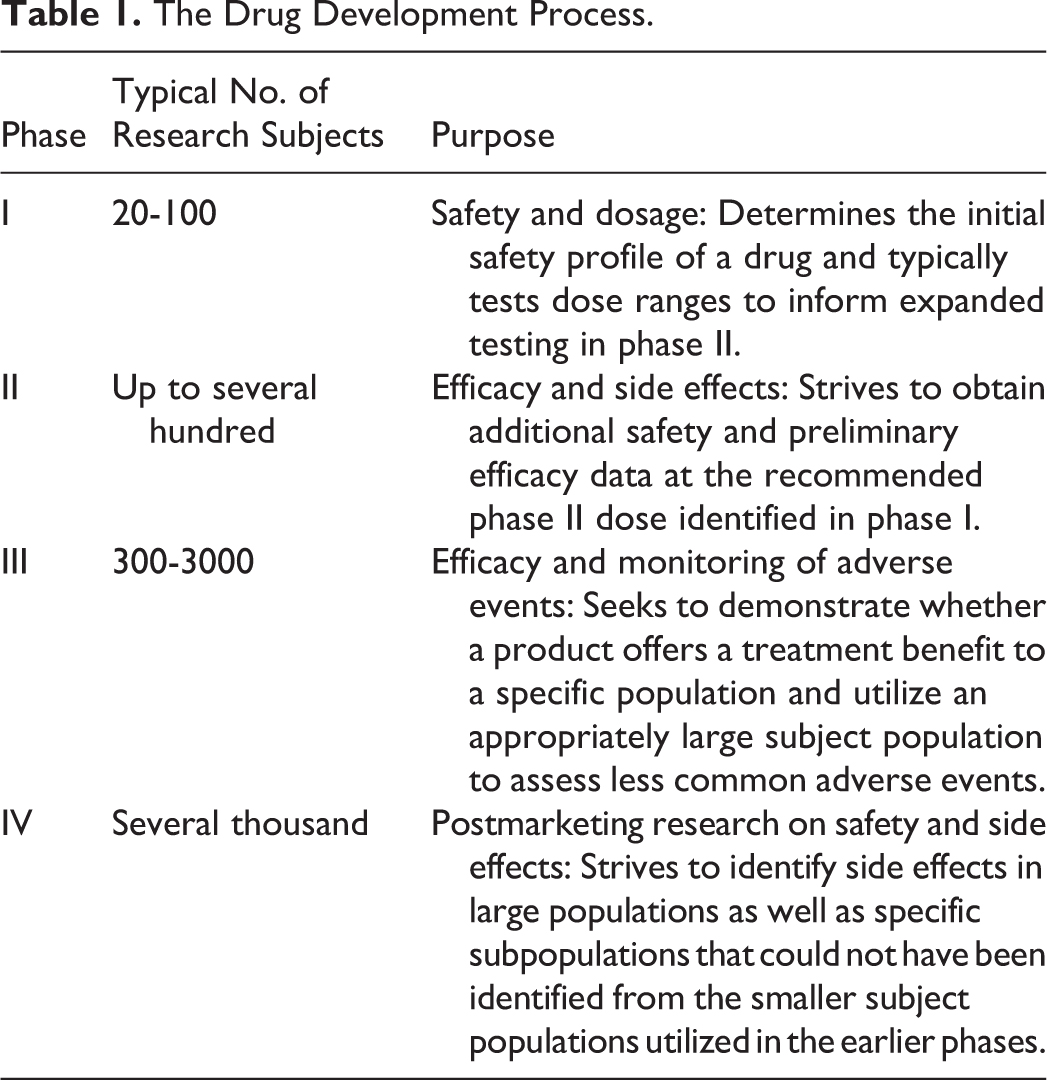
Clinical trials are designed in 4 phases, each intended to generate a specific type of data (Table 1 4 ). Early phase studies (I and II) focus on safety, while phase III shifts its focus to efficacy. Marketing approval is sought from FDA after phase III. Phase IV involves postmarketing research to identify side effects in large populations as well as specific subpopulations, such as age groups, health status, ethnicity, and so on.
The Drug Development Process.
Ongoing Revisions to NIH Guidelines and the Role of the RAC
The NIH Recombinant DNA Advisory Committee was established in 1974 to advise the NIH director on matters pertaining to the conduct and oversight of research involving recombinant DNA molecules. A 1982 report by the President’s Commission titled Splicing Life proposed changes to oversight of recombinant DNA research. 5 In response to that report, the RAC issued the first version of Points to Consider in its 1985 report, “Design and Submission of Human Somatic-Cell Gene Therapy Protocols” and the RAC’s mission was temporarily expanded to encompass review of gene therapy research, referred to as clinical trials involving human gene transfer (HGT) in NIH Guidelines. 6
The RAC was not intended to serve as the sole entity for review of gene therapy studies, and in 1995, the NIH and FDA outlined an agreement where both agencies would jointly determine which research protocols warranted public RAC review. In 1997, NIH Guidelines shifted the focus of the RAC to an advisory role with the FDA assuming sole authority to approve gene transfer protocols as well as gene therapy products.
In 2012, the American Society of Gene and Cell Therapy (ASGCT) submitted a letter to the RAC requesting the elimination of the RAC review requirement for gene therapy studies. In the letter, ASGCT pointed out that the major concerns that lead to the requirement for RAC review (modification of the germline and creation of novel pathogen vectors) have not presented themselves in clinical trials. ASGCT argued that research has created a sufficient body of knowledge to assess the risks of gene therapy research without RAC review. Furthermore, the requirements for FDA review as well as IRB and IBC review made the RAC review duplicative and time-consuming and required a level of review not afforded to other areas of clinical research. In 2013, a panel of gene therapy experts convened at the Institute of Medicine to reevaluate the gene therapy field and the role of the RAC. The panel’s report suggested streamlining the RAC review process and reserving RAC review to novel technologies and studies with risks that are difficult to assess.
The April 2016 version of NIH Guidelines, which were issued in part to evolve the process for RAC review, instructed the local oversight bodies (ie, the IRB and IBC) at an institution (ie, the research site) to determine whether to recommend RAC review. That version of the guidelines provided 3 criteria by which RAC review would be warranted. These criteria largely focused on the novelty of the science and the complexities involved in the risk assessment or possible toxicities. RAC review was deemed to be warranted if any of the following applied
7
:
the protocol uses a new vector, genetic material, or delivery methodology that represents a first-in-human experience, thus presenting an unknown risk; or the protocol relies on preclinical safety data that were obtained using a new preclinical model system of unknown and unconfirmed value; or the proposed vector, gene construct, or method of delivery is associated with possible toxicities that are not widely known and may render it difficult for oversight and federal regulatory bodies to evaluate the protocol rigorously.
Furthermore, the NIH director retained the ability to select a protocol for RAC review if it raised significant scientific, societal, or ethical concerns.
Regardless of the determination made by the IBC and IRB, the April 2016 NIH Guidelines required gene therapy studies to register with the NIH Office of Science Policy (OSP). The registration process served as confirmation that RAC review was not warranted. NIH Guidelines allotted NIH OSP 10 working days to acknowledge the completion of the registration, which then allowed the IBC to issue final approval.
Problems with the HGT Review Process
The process outlined in the 2016 version of NIH Guidelines called for 2-step IBC review bookending a 2-week review by the NIH. At an institution with monthly IBC meetings, review of HGT studies could last 2 to 3 months, or longer, especially if monthly submission deadlines were missed. Further delays would be incurred if the study were selected for review at a quarterly RAC meeting. While this process was an improvement over requiring RAC review for all gene therapy studies, it was inefficient. However, this process was an evolutionary step allowing NIH OSP to reassess review of gene therapy studies. While the registration process took place under the April 2016 version of NIH Guidelines, only 3 of 275 HGT studies were determined to require RAC review. 8
The Continuing Evolution of the Process
On August 16, 2018, the NIH proposed revisions to NIH Guidelines that focused predominantly on review of human gene transfer studies. The revised NIH Guidelines, which were published on April 25, 2019, lack an appendix devoted to review of HGT studies, thereby eliminating the protocol submission, review, and reporting requirements. The new process for review of gene therapy studies is similar to the review process under the vaccine exemption previously under Appendix M-III-A of the 2016 NIH Guidelines. That exemption applied to recombinant vaccines against microbial immunogens delivered with vectors that do not persist in the research subject. The risks were considered to be well understood, and involvement of the RAC was not required since adequate oversight was provided by the FDA, IRB, and IBC.
While the revised NIH Guidelines lack an appendix addressing human gene transfer, guidance is available to IBCs in the form of Points to Consider: Institutional Biosafety Committee (IBC) Review of Human Gene Transfer Protocols. The guidance document lists the recommended documents for review of HGT studies previously listed in Appendix M-I when it was devoted to HGT studies. Furthermore, the document outlines the roles of the IBC in review of HGT studies as outlined in Section IV-B of NIH Guidelines. 9
On the day the comment period opened for what was at the time the proposed revisions, NIH Director Francis Collins issued a statement that “NIH…intends to restore the NIH Recombinant DNA Advisory Committee (RAC), which has long played a role in advising the NIH director on gene therapy, to its original vision by proposing it focus on the scientific, safety, and ethical issues associated with new and emerging biotechnologies.” 10 To further emphasize the more general role of the RAC, the committee has been renamed the Novel and Exceptional Technology and Research Advisory Committee (NExTRAC). The committee’s amended charter charges it with advising the NIH director “on matters related to the conduct and oversight of research involving emerging technologies in biomedical science (also referred to as emerging biotechnologies).” 11 The NIH FAQ on the April 2019 Amendment of the NIH Guidelines mentions the scope of the committee is “not necessarily limited to recombinant or synthetic nucleic acid molecule research.” 12
Removing the requirement for submission, review, and reporting to the NIH OSP and associated RAC review is a welcome change to many in the clinical research field who saw the RAC registration and review requirement as a delay with the potential to turn into an extended delay (if a study was chosen for RAC review at a quarterly meeting).
Many in the biosafety field have expressed concern about decreasing the role of the RAC and possibly even eliminating the RAC altogether. But elimination of the RAC was not included in the proposed NIH Guidelines revisions; the RAC in the form of the NExTRAC remains in its advisory capacity to the NIH director in matters pertaining to emerging biotechnology. Removing the RAC from the review process for gene therapy studies is due to the wealth of knowledge currently available about the technology and its associated risks. At a March 6, 2019, presentation at the University of Minnesota, Associate Director of NIH OSP Carrie Wolinetz stated, “When devising policy for gene therapy, we need to stop thinking about gene therapy as an emerging technology. Gene therapy has emerged.” 13
The NExTRAC is now unencumbered by review of large volumes of HGT studies and can focus on emerging biotechnology requiring the focus of a national panel of experts. Many issues are likely on the horizon for the NExTRAC, such as emerging developments in gene editing technology, especially as they pertain to the application and delivery to human research subjects. Clinical application of genetic engineering technology to matters pertaining to human reproduction including embryonic development and editing of the germline are likely hot button issues.14 –17 Environmental issues such as gene drives and environmental release are also likely matters of future discussion.
FDA Leads the Charge into the Future of Gene Therapy
Since the FDA is tasked with assessing the safety and efficacy of therapeutic products, it might be a natural progression for the oversight burden for gene therapy products to transition from the NIH to the FDA. The FDA has taken a number of steps in recent years to assist in bringing gene therapy from the realm of research to the world of approved and licensed therapeutics for clinical use:
The FDA has issued guidance documents for the manufacture of gene therapy products and the design of clinical trials (Table 2).
18
The 21st Century Cures Act authorized the FDA to create the Regenerative Medicine and Advanced Therapies (RMAT) designation to allow for expedited review of regenerative medicines and advanced therapies. The associated guidance issued by the FDA stated the RMAT designation also applies to “gene therapies, including genetically modified cells, that lead to a durable modification of cells or tissues.”
19
The FDA is expanding its capabilities to review gene therapy studies to accommodate the growing field. In a June 2018 interview, FDA Commissioner Scott Gottlieb disclosed that he expects the agency to have approved 40 gene therapies by 2022, a gargantuan number considering only 4 approvals had been issued at the time.
20
In a statement issued in January 2019, Gottlieb and CBER Director Peter Marks mentioned plans for:
hiring 50 additional clinical reviewers for cell and gene therapy 200 IND applications for gene therapy products submitted per year by 2020 10 to 20 gene therapy approvals per year by 2025.
21
FDA-Issued Gene Therapy Guidance Documents.

In the joint statement, Gottlieb and Marks write that the growth in gene therapy “reflects a turning point in the development of these technologies and their application to human health…. It’s similar to the period marking an acceleration in the development of antibody drugs in the late 1990s, and the mainstreaming of monoclonal antibodies as the backbone of modern treatment regimens.” 21
Conclusion
The oversight structure for recombinant DNA has been evolving since its inception in the 1970s. While gene therapy was once a highly experimental science fraught with unknowns, it is now a well-established field. The gene therapy field has recently produced its first FDA-approved therapeutics and has a pipeline of other investigational products in the final stages of clinical trials before they can be evaluated by the FDA as safe and effective therapeutics. As research continues to evolve, so must the oversight structure. Biosafety professionals and IBCs have always played key roles in contributing to the safe, evidence-based advancement of gene therapy research. With the recent regulatory changes and current surge in gene therapy research, the importance of those roles has increased dramatically.
Footnotes
Acknowledgments
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. Daniel Eisenman is employed by Advarra as Director of Biosafety Services.