
Abstract
All disinfectants and pesticides marketed for use in United States must meet safety requirements as described in OCSPP 810.2200. The Antimicrobial Testing Program (ATP) ensures that the Environmental Protection Agency (EPA) approved hospital disinfectants and tuberculocides in the marketplace continue to meet stringent efficacy standards. This paper addresses the issues of getting a hospital disinfectant approved by the EPA, some of the challenges encountered and results achieved for a number of hospital related pathogens including MRSA, Pseudomonas sp., H1N1, etc. using a novel approach. The paper also addresses the differences between hands-free and wipes technologies, sprays, and their approval pathways. The paper presents the results from Good Laboratory Practice Studies (GLP) submitted to EPA for a novel technology using The SteraMist™ Surface and Environmental systems. SteraMist™ Binary Ionization Technology® (BIT™), (TOMI, Beverly Hills, CA), converts a 7.8% hydrogen peroxide solution into a Hydroxyl Radical mist. This EPA registered solution is passed through an atmospheric cold plasma arc where activation occurs. Activation creates a mist/fog containing a high concentration of Reactive Oxygen Species (ROS), mainly the Hydroxyl Radical. Nine human pathogens are discussed including Gram positive and negative bacteria, virus species and mycobacterium. The mist/fog referred to as Activated Ionized Hydrogen Peroxide (AIHP) is delivered via a handheld application system or a standalone computerized environmental system.
Keywords
Introduction
All disinfectants and pesticides marketed for use in United States must meet safety requirements as described in OCSPP 810.2200, 1 Applicability. This guideline describes test methods that EPA believes will generally satisfy testing requirements of the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA) (7 U.S.C. 136, et seq.) and the Federal Food, Drug, and Cosmetic Act (FFDCA) (21 U.S.C. 346a). It addresses testing to demonstrate the effectiveness of antimicrobial pesticides bearing claims as disinfectants, fungicides, virucides, and tuberculocides. (EPA 712-C-07-074)
This paper will demonstrate the process a company must follow to register a product and will demonstrate the stringency of performance encountered. required. The paper will review studies performed to clear a manufacturer’s products through EPA using the current statutes. The author hopes to make the reader and user of hospital grade disinfectant products more aware as to the differences between claims made by manufacturers and distributors. This knowledge will potentially impact on their use of these products in a hospital or healthcare setting.
The Environmental Protection Agency (EPA) is charged with monitoring an antimicrobial’s efficacy and every company must submit their product for approval before marketing the product or making claims about the product’s efficacy as a hospital grade disinfectant The EPA rules apply to liquids, mist/fogs and wipes.
The EPA program is described as follows (section 40 CFR, 160) 4 .
Pesticide Registration
An overview of the registration program is available at https://www.epa.gov/pesticide-registration/antimicrobial-testing-program#overview 3 . In summary, the EPA Antimicrobial Testing Program evaluates the product claims for disinfectant products. Only products that meet stringent testing requirements can be registered with the EPA and marketed using terms such as “hospital disinfectant” or “tuberculocidal”. Registration of a disinfectant for a specific use requires submission of data that demonstrates efficacy using standardized procedures. The testing must be conducted using Good Laboratory Practices (GLP) (https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/epa/epa_glp40_160.pdf) and (https://www.fda.gov/ohrms/dockets/98fr/980335s1.pdf 21 CFR Part 58)4,5 and must follow the appropriate study standard, OCSPP 810.2200 (Office of Chemical Safety and Pollution Prevention), etc. 1 See Table 1, Disinfectant for Use on Hard Surfaces-Efficacy Data recommendations. The nature of the testing varies as to the whether the product is a liquid, wipe, spray, or a fog/mist. The GLP studies and protocols are designed to meet the above standards and must be approved by the EPA before starting.
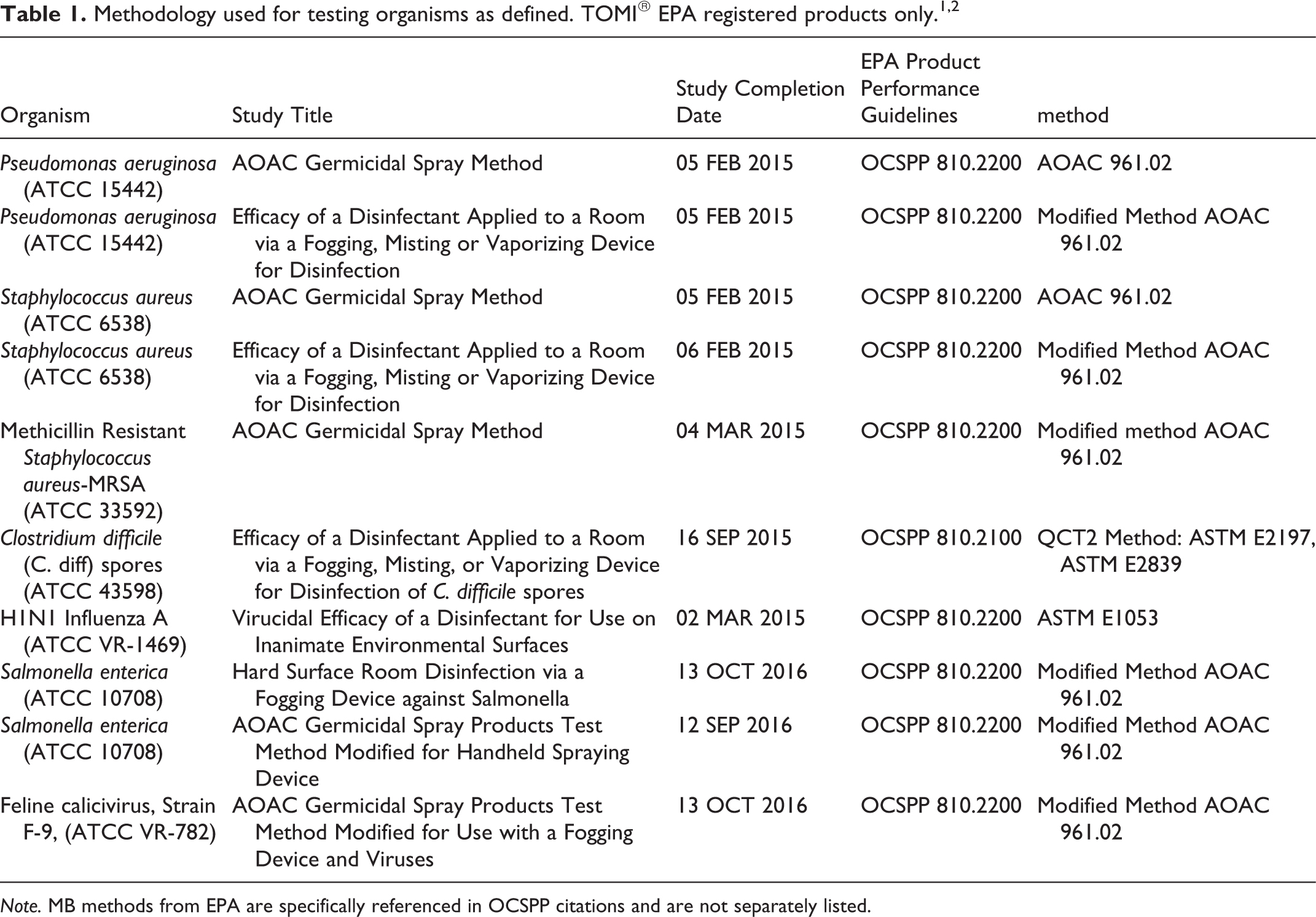
Note. MB methods from EPA are specifically referenced in OCSPP citations and are not separately listed.
If approved, the primary product will be issued an EPA registration number that must appear on any label listing disinfectant claims. A second number may appear on the label—this indicates a registered “supplemental distributor product” based on the primary product. For example, EPA registration Number 12345-12 is the primary registration number; EPA registration number 12345-12-2567 refers to a distributor product.
Updates to Registration Procedures
In April, 2013, the EPA issued a certified letter to all companies with registered antimicrobial products. In 2013, the Sterimist product only had fungal EPA claims Reg. No. 90150.1 at that time. All other studies were performed at a later date as shown in Table 1.
“As explained in the letter, we are concerned that fogging/misting products may not be as effective as claimed, and we want to ensure that these fogging/misting products are accurately labeled. By this letter, we are asking companies either to provide existing efficacy data, or to commit to provide new data, that address the public health claims for the fogger/mister products. Alternatively, the registrant may elect to revise the labeling of the affected products and registrations to delete the public health claims for the fogger/mister products.” “The Agency now considers the claims made for disinfection and sanitization for products applied by fogging/misting to be public health-related. This would include fogging/misting products that bear label claims that the product can be used as ‘‘an adjunct”‘ to sanitization or disinfection.” “The fear is that the differences in particle size may affect the amount of active ingredient on the surface. “A surface treated by fogging/misting does not receive the same amount of active ingredient per unit area as the standard methods of application and, as a result, the level of efficacy actually achieved may not be the same level claimed on the label.”
6
EPA Sept 19, 2016 report 16-p 0316, states that the memorandum is in response to the Office of Inspector General’s (OIG) Draft Report Entitled “EPA Needs a Risk-Based Strategy to Assure Continued Effectiveness of Hospital-Level Disinfectants.”
The product data presented demonstrates the efficacy of the required log kill or other stated EPA criteria for the TOMI Environmental Solutions', Inc. SteraMist™ BIT™ systems for Staphylococcus aureus, Methicillin Resistant Staph aureus (MRSA), Pseudomonas aeruginosa, Influenza A (H1N1), Clostridium difficile spores, Salmonella enterica, Geobacillus stearothermophilus, and Mycobacterium bovis. Additionally, data for Norovirus is presented which has recently cleared EPA (July 2017).
The studies presented here are only for classification of the SteraMist™ BIT™ system as a hospital disinfectant. They do not represent the Food and Drug Agency (FDA) requirements for high level disinfection and those studies are not considered in this work. The disinfectant system as used in these studies is not classified as a medical device and is not regulated as such.
Materials and Methods
Companies using the dip test for spray and wipe technologies must follow the guidelines found In the Official Methods of Analysis of the AOAC International, Chapter 6, Disinfectants, Use-dilution methods (955.14, 955.15, & 964.02). 8 In these studies carriers are inoculated with specific organisms with three different lots of disinfectant, one lot being older than 60 days old; 60 carriers per organism used, with 59/60 carriers being negative for the organism after disinfectant treatment. In summary, carriers are inoculated with challenge organism and fully submersed in the disinfectant for the time listed on the label, at which time carriers are removed, neutralized, incubated, and reviewed for growth. The test is also run where the challenge organism is put on the carrier, and then the disinfectant solution is placed on the carrier via pipette, immersing the carrier/inoculation site. Details are found in cited guideline and were not used by the GLP laboratories in testing the SteraMistTM Bit Solution system(s) as the studies done for the spray/fog systems use different methodologies compared to wipe technologies as detailed in Table 1 methods.
A novel technology using The SteraMistTM Surface and Environmental systems, SteraMist™ Binary Ionization Technology® (BIT™), (TOMI®, Beverly Hills, CA), converts a 7.8% hydrogen peroxide solution into a hydroxyl radical mist. This EPA registered solution is passed through an atmospheric cold plasma arc where activation occurs. Activation creates a mist/fog containing a high concentration of reactive oxygen species (ROS), mainly the hydroxyl radical (OH-). The mist/fog referred to as Activated Ionized Hydrogen Peroxide (AIHP) is delivered via a handheld application system or a standalone environmental system with the same properties monitored by robotic software using three units identical to the handheld units but placed on tripods.
All studies presented in the paper were performed for TOMI by contracted laboratories meeting the GLP requirements as defined in 40 CFR Part 160 and substance characterization as defined in Subpart F (160.105) 4 that apply to studies for determination of disinfection on hard, non-porous surfaces. A sponsor may not conduct the study. The methods used to do the studies are defined by The Association of Official Analytical Chemicals (AOAC) recommended tests and must be performed as written (OCSPP Test guideline 810.2000 and 810.2100 for general testing considerations must be met prior to initiating test). Table 1 1,8 and any modifications must be approved by the EPA prior to start of the study. Hospital or healthcare disinfectant/hard non-porous surfaces spray products must use Staphylococcus aureus (SA) (ATCC 6538) and Pseudomonas aeruginosa (PA) (ATCC 15442) as part of their testing for acceptance by EPA. For viruses, the protocol was modified for viruses to include a chemical neutralizer per ASTM E1053. This standard was used for viruses as claimed on the label. In this study H1N1 Influenza A (ATCC VR-1469) and Norovirus surrogate feline calcivirus was tested. Additionally, S. aureus (MRSA, ATCC 6538), Clostridium difficile (C. diff) spores (ATCC 43598), Salmonella enterica (ATCC 10708), Geobacillus stearothermophilus (ATCC 12980), and Mycobacterium bovis (BCG) were tested.
Methods (see Table 1 for specific protocols as defined by EPA requirements)
Handheld Unit
The GLP laboratory followed established protocol as defined on pages 3-4 of protocol TEST01122314 GS. for SA and PA
9
; Protocol TES01120614 GS for MRSA pages 8-10
10
; Protocol TES01120614.FLU A for H1N1 pages 3-12 modifications included
11
; Protocol Number P1619 pages 25-38 for Salmonella enterica and protocol modifications page10
12
; and All lots of BIT™ Solution being used had to meet acceptance criteria as defined by the manufacturer and accompanied with a certificate of analysis. The Solution is “ready to use” and does not require dilution. For the handheld unit, for each bacterial and viral organism, the cultures were grown per protocol and coated onto carriers, usually glass microscope slides. Carriers were inoculated, and dried per protocols. (ref above) Three lots of BIT™ Solution were tested for efficacy for each organism as noted above. In testing Norovirus, protocols required maintenance of 4-log virus on control carriers during trial and a minimum of BIT™ Solution was applied to the carriers using the SteraMist™ Surface Unit for 5 seconds/ 0.0929 m2 (1 ft2) at 24 inches and held at 16.7-27% relative humidity for 7 minutes at room temperature 20.9 - 21.8 degrees C depending on organism being tested by GLP laboratory after a trigger release of two seconds. The carriers were transferred using sterile technique to neutralizing solution. All controls were tested according to protocols9,10,13–15 as detailed below and the success of the testing was evaluated by the following parameters: The initial carrier numbers control enumerations must demonstrate a geometric mean density ≥ 1 x 105 CFU/carrier unless the product fails, in which case the geometric mean may be lower. The final carrier numbers control enumerations must demonstrate a geometric mean density ≥ 1 x 104 CFU/carrier unless the product fails, in which case the geometric mean may be lower. In the event that the final numbers control is lower than that specified above, the mean density of control substance treated carriers (10 carriers treated with control substance) may be used in place of the final carrier numbers control. The control substance treated carrier enumerations must demonstrate a geometric mean density ≥ 1 x 104 CFU/carrier unless the product fails, in which case the geometric mean may be lower. The subculture/neutralization sterility control test tube is negative for growth. Both of the viability growth control test tubes are positive for growth. * The neutralization control subculture/neutralization test tube is positive for growth. *The neutralization control inoculum demonstrates between 10 and 100 CFU. Carrier sterility control tube is negative for growth. Test microorganism purity control demonstrates growth and lack of contaminant species.
*The neutralization control data is not shown as the criteria were met as specified in parts e and f. If the criteria had not been met, the study would have failed.
Environmental Unit
Protocol TES01062615.RDT pages 8-16 for C. difficile
14
; Protocol TES01030716.RDT page 3 for Mycobacterium bovis
15
; Protocol MRID 488313-03 (field study) pages 2-4 for Geobacillus stearothermophilus
16
; Protocols NG7535 and GLP 1507 for Norovirus.
13
In the case of the C. diff spores these were tested using the SteraMist™ Environment System and carriers were systematically placed in a location according to room dimension as described on page 26 of protocol.TES01062615.RDT.
14
and Table 1. The BIT™ Solution for C. diff spores was tested in a 103.74 m3 (3663.7 Ft3) room at a dosage of 0.5 ml /.0283 m3(0.5 ml/ Ft3.) with a 15 minute contact time and a 26-29 minute aeration time. Mycobacterium bovis was tested with the SteraMist™ Environment System, but did not follow GLP protocols per sponsor.
15
The data represented here were feasibility studies and GLP studies are now being undertaken and are in progress. Briefly, the 20 inoculated glass carriers were placed at diverse locations in a sealed testing room. They were fogged for 23 minutes, with a 17 minute dwell time, followed by 50 minute aeration time. All appropriate sterility, purity, and recovery controls were included. For the Field Study for Geobacillus stearothermophilus (ATCC 12980)
16
62 inoculated stainless steel carriers were placed in a sealed, 103.74 m3 (3663.7 ft3) room and fogged for 24 minutes, with a 15 minute dwell and 90 minute aeration time. Three lots of BIT™ solution > 60 days old were used. Carriers were transferred in a sterile manner and tested for growth per protocol. The SteraMist™ Environment System was positioned within the enclosure.
Any deviations from protocols were recorded for all the above studies. All controls were tested according to protocols as with handheld unit above.
Results
Criteria of acceptability as a “hospital disinfectant” are defined for each organism as it varies as to requirements per “accepted” EPA protocol used by the GLP laboratory.
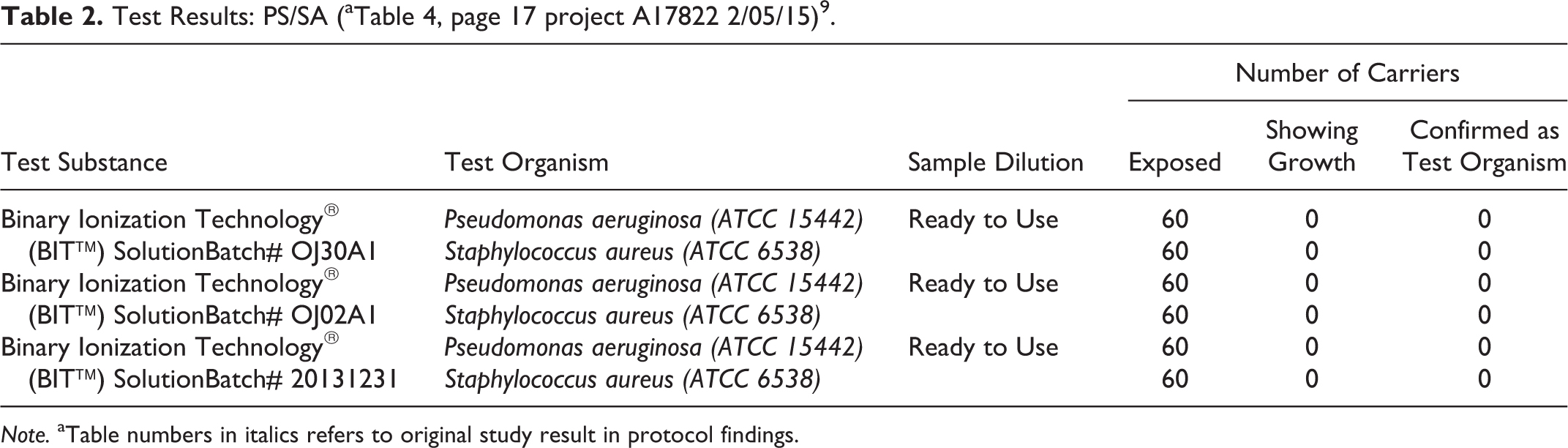
Note. aTable numbers in italics refers to original study result in protocol findings.
2.
3.
The test substance must demonstrate a minimum of 6 Log10 reduction in numbers of test organism as compared to the carrier population control (post testing).
Controls must perform according to protocol criteria. (Table 3).
All lots of solution must have color change indicator present.

*Chemical indicator results were matched with 3 carriers for each lot of BIT™ solution: Carriers 4, 5, and 7 each had color change present validating the results.
Note. aTable numbers in italics refers to original study result in protocol findings.
4.
A valid test requires that at least a 4 Log10 of infectivity be recovered from the dried virus control film; That when cytotoxicity is evident, at least a 3 Log10 reduction in titer is demonstrated beyond the cytotoxic level; That the cell control be negative for infectivity. Note: an efficacious product must demonstrate complete inactivation of the virus at all dilutions.
H1N1 Test results: Effects of Binary Ionization Technology® (BIT™) Solution (Batch# OJ30A1 & Batch# OJ02A1) Following a 5 second application per 0.0929 m2 (1 Ft 2) time and 7 minute contact time to Influenza A (H1N1) Virus dried on an Inanimate Surface. 12
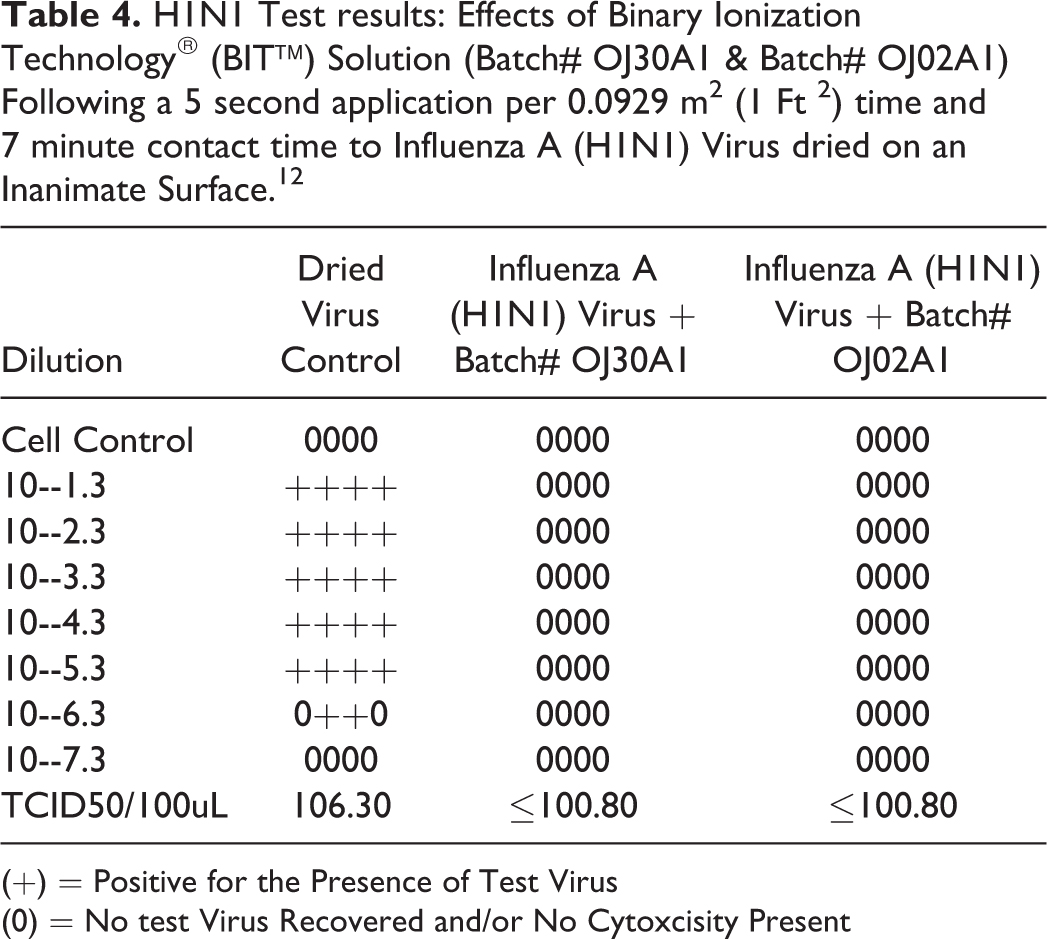
(+) = Positive for the Presence of Test Virus
(0) = No test Virus Recovered and/or No Cytoxcisity Present
5.
All controls had appropriate results. Conclusion: 60/60 carriers were disinfected within 7 minutes +/- 5 seconds using three lots of BIT™ Solution (Table 5a, b, c).
Test Results: a,b,c Carrier Enumeration for Salmonella enterica tested against 3-lots of BITTM Solution. 15
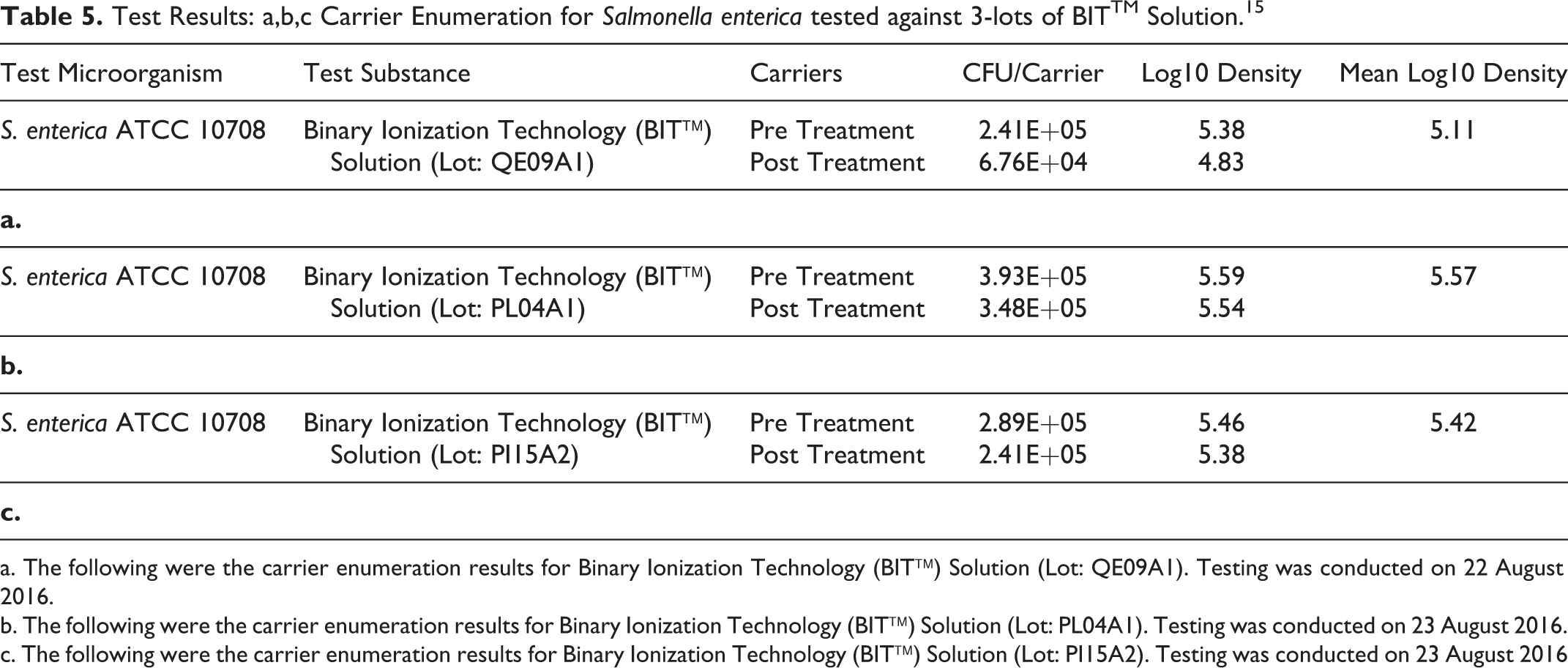
a. The following were the carrier enumeration results for Binary Ionization Technology (BIT™) Solution (Lot: QE09A1). Testing was conducted on 22 August 2016.
b. The following were the carrier enumeration results for Binary Ionization Technology (BIT™) Solution (Lot: PL04A1). Testing was conducted on 23 August 2016.
c. The following were the carrier enumeration results for Binary Ionization Technology (BIT™) Solution (Lot: PI15A2). Testing was conducted on 23 August 2016
6.
Mycobacterium bovis Carrier population control results. 13

CFU = Colony Forming Unit.
7.
G. stearothermophilus Carrier Results. 14

8.
A minimum of 4 log10 reduction infectious viruses are recovered from the virus control carrier. Viral cytopathic effects are distinguishable from cytotoxic effects caused by test substance exposure. Neutralization effectiveness is demonstrated by recovery of comparable levels of infectious viruses from control. Assay wells designated as sterility controls are absent of infectivity, contamination, and cytotoxicity.
Passing criteria as noted before includes complete inactivation of test virus at all dilutions with a minimum of
Test Results Norovirus Surrogate, NG7535. 16
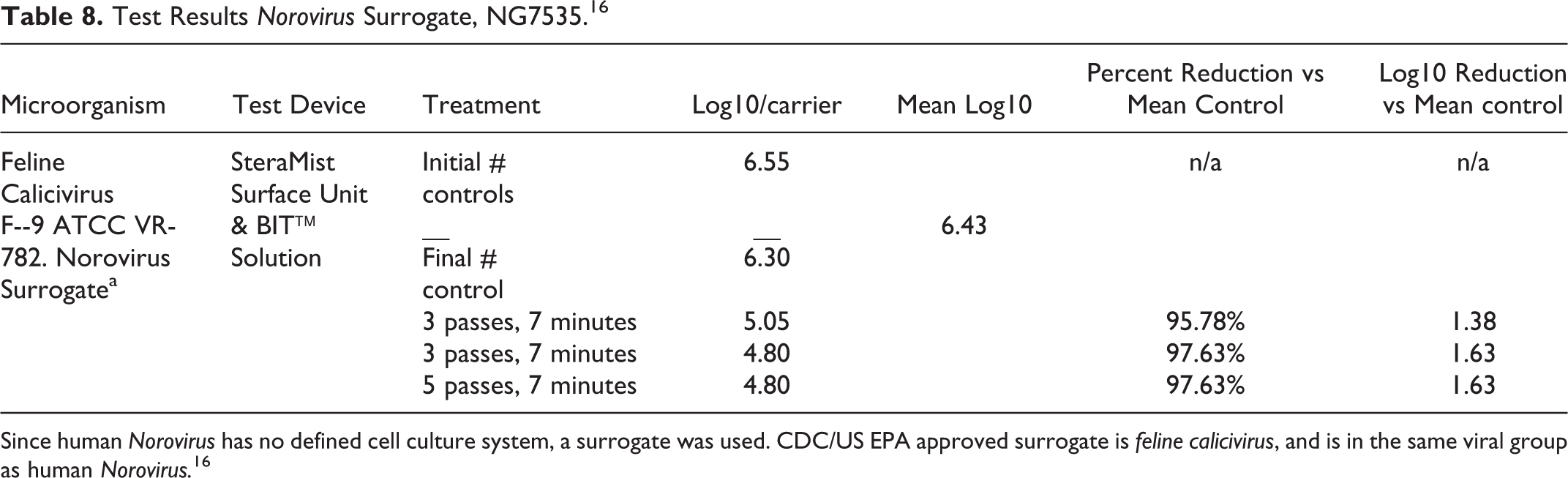
Since human Norovirus has no defined cell culture system, a surrogate was used. CDC/US EPA approved surrogate is feline calicivirus, and is in the same viral group as human Norovirus. 16
The following is a summary of the results gathered from the test substance lots evaluated, as well as a statement of their ability to meet the U.S. EPA Product Performance Guidelines for Disinfectants for Use on Hard Surfaces outlined in OCSPP 810.2200 and the success criteria detailed in the approved Protocol GLP 1507, MicroChem. Test substance Binary Ionization Technology (BIT) Solution (Lot: QE09A1) met the U.S. EPA Product Performance Guidelines for Disinfectants for Use on Hard Surfaces outlined in OCSPP 810.2200 and the success criteria detailed in the approved protocol against Feline calicivirus, Strain F-9, ATCC VR-782 at the reported exposure times. No infectious viral particles were recovered from any of the locations tested with this lot of test substance. Test substance Binary Ionization Technology (BIT) Solution (Lot: PL04A1) met the U.S. EPA Product Performance Guidelines for Disinfectants for Use on Hard Surfaces outlined in OCSPP 810.2200 and the success criteria detailed in the approved protocol against Feline calicivirus, Strain F-9, ATCC VR-782 at the reported exposure times. No infectious viral particles were recovered from any of the locations tested with this lot of test substance.
Discussion
GLP studies are highly structured and must meet the standards as defined and listed in this paper. There is little to no room for modification and the substance being tested must meet the criteria as stated for efficacy and approved by the EPA. These claims must be reflected in the product labeling. The stringent requirements of the GLP studies may not reflect the “actual use” of the product in a hospital or a healthcare setting as healthcare settings are not GLP controlled environments. The end-users of the technology do not generally have the expertise or the resources to do the necessary testing to support the claims of the manufacturers as demonstrated by the GLP studies. But, the studies do give the user confidence that the claims made by the manufacturer are in fact supported by independent GLP laboratories and reviewed for efficacy of log kill by the EPA. Additionally, the previously registered disinfectants may now be subject to ongoing re-testing as required by recent EPA actions 8 to further assure product continued efficacy.
The above studies demonstrate the efficacy of both the SteraMist™ handheld Surface Unit and the SteraMist™ Environment System with BIT™ Solution for a large number of clinically significant organisms. These studies are very difficult to perform as compared to the dip test technology where carriers are simply immersed in the disinfectant solution for a period of time. These studies are the first reported studies to the authors knowledge that demonstrate the combination of a disinfection solution and the effect of the dispersal method with the resulting effect on the killing efficacy for multiple organisms as now required by EPA. 6
The GLP and other feasibility studies presented in this report have demonstrated the effectiveness of kill for Gram positive and Gram negative bacteria; spore forming organisms such as, C. difficile and Geobacillus stearothermophilus; Acid Fast Bacilli, Mycobacterium sp.; and viruses Influenza A-H1N1, and Norovirus with the combination of TOMI™ BIT™ Solution and cold plasma arc dispersal system whether handheld or as an environmental robot controlled system. The studies presented have also demonstrated the rigorous process of EPA registration for a product to be classified as a hospital disinfectant.
Footnotes
Declaration of Conflicting Interests
The author reports she is on the scientific advisory board of TOMI, Beverly Hills, CA.
Funding
The author received financial support from TOMI for the research, authorship, and publication of this article.