
Abstract
Huntington’s disease, like other neurodegenerative diseases, continues to lack an effective cure. Current treatments that address early symptoms ultimately fail Huntington’s disease patients and their families, with the disease typically being fatal within 10–15 years from onset. Huntington’s disease is an inherited disorder with motor and mental impairment, and is associated with the genetic expansion of a CAG codon repeat encoding a polyglutamine-segment-containing protein called huntingtin. These Huntington’s disease mutations cause misfolding and aggregation of fragments of the mutant huntingtin protein, thereby likely contributing to disease toxicity through a combination of gain-of-toxic-function for the misfolded aggregates and a loss of function from sequestration of huntingtin and other proteins. As with other amyloid diseases, the mutant protein forms non-native fibrillar structures, which in Huntington’s disease are found within patients’ neurons. The intracellular deposits are associated with dysregulation of vital processes, and inter-neuronal transport of aggregates may contribute to disease progression. However, a molecular understanding of these aggregates and their detrimental effects has been frustrated by insufficient structural data on the misfolded protein state. In this review, we examine recent developments in the structural biology of polyglutamine-expanded huntingtin fragments, and especially the contributions enabled by advances in solid-state nuclear magnetic resonance spectroscopy. We summarize and discuss our current structural understanding of the huntingtin deposits and how this information furthers our understanding of the misfolding mechanism and disease toxicity mechanisms.
Impact statement
Many incurable neurodegenerative disorders are associated with, and potentially caused by, the amyloidogenic misfolding and aggregation of proteins. Usually, complex genetic and behavioral factors dictate disease risk and age of onset. Due to its principally mono-genic origin, which strongly predicts the age-of-onset by the extent of CAG repeat expansion, Huntington’s disease (HD) presents a unique opportunity to dissect the underlying disease-causing processes in molecular detail. Yet, until recently, the mutant huntingtin protein with its expanded polyglutamine domain has resisted structural study at the atomic level. We present here a review of recent developments in HD structural biology, facilitated by breakthrough data from solid-state NMR spectroscopy, electron microscopy, and complementary methods. The misfolded structures of the fibrillar proteins inform our mechanistic understanding of the disease-causing molecular processes in HD, other CAG repeat expansion disorders, and, more generally, protein deposition disease.
Introduction
Huntington’s disease (HD) is an inherited neurodegenerative disease (NDD) in which the mutated protein undergoes misfolding and aggregation in patients’ neuronal cells. As such, it is one example of an expanding class of protein misfolding and deposition diseases that include Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). 1 The nature of the disease-causing mutation makes HD the most well-known example of a family of CAG repeat expansion disorders. In each of these disorders, the mutation affects a different gene with a naturally occurring CAG repeat, which, when expanded past a disease-specific threshold length, results in an age-dependent NDD (Figure 1(a)). On the protein level, this CAG repeat translates into a polyglutamine repeat, which in the wild-type proteins is typically between 10 and 35 glutamines long. Although the mutated proteins are involved in divergent functional roles, and include both soluble and membrane-bound proteins, the diseases share common features. Six CAG repeat expansion diseases are classified as spinocerebellar ataxias (SCAs) with cerebellum atrophy leading to symptoms such as poor hand or speech coordination, eye movement, or cognitive impairment. Dentatorubral-pallidoluysian atrophy (DRPLA) is characterized by dementia, ataxia, and choreoathetosis. Thus, a common phenotype seems independent of the functions of the mutated protein, which could be rationalized by commonalities in disease-mechanisms driven by protein-based gain-of-toxic-function in these and other protein misfolding diseases, 1 rather than merely a loss of the native function. In these NDDs, amyloid formation is a common hallmark, reflecting protein aggregation accompanied by a conformational change of the affected protein to a characteristic β-sheet architecture. 1 In AD and PD, our understanding of this conformational transition was recently boosted by high-resolution structures of protein filaments.2–4 Such structural information on the misfolded protein assemblies has proved more challenging to obtain in the case of HD, but important recent progress will be examined in this review.
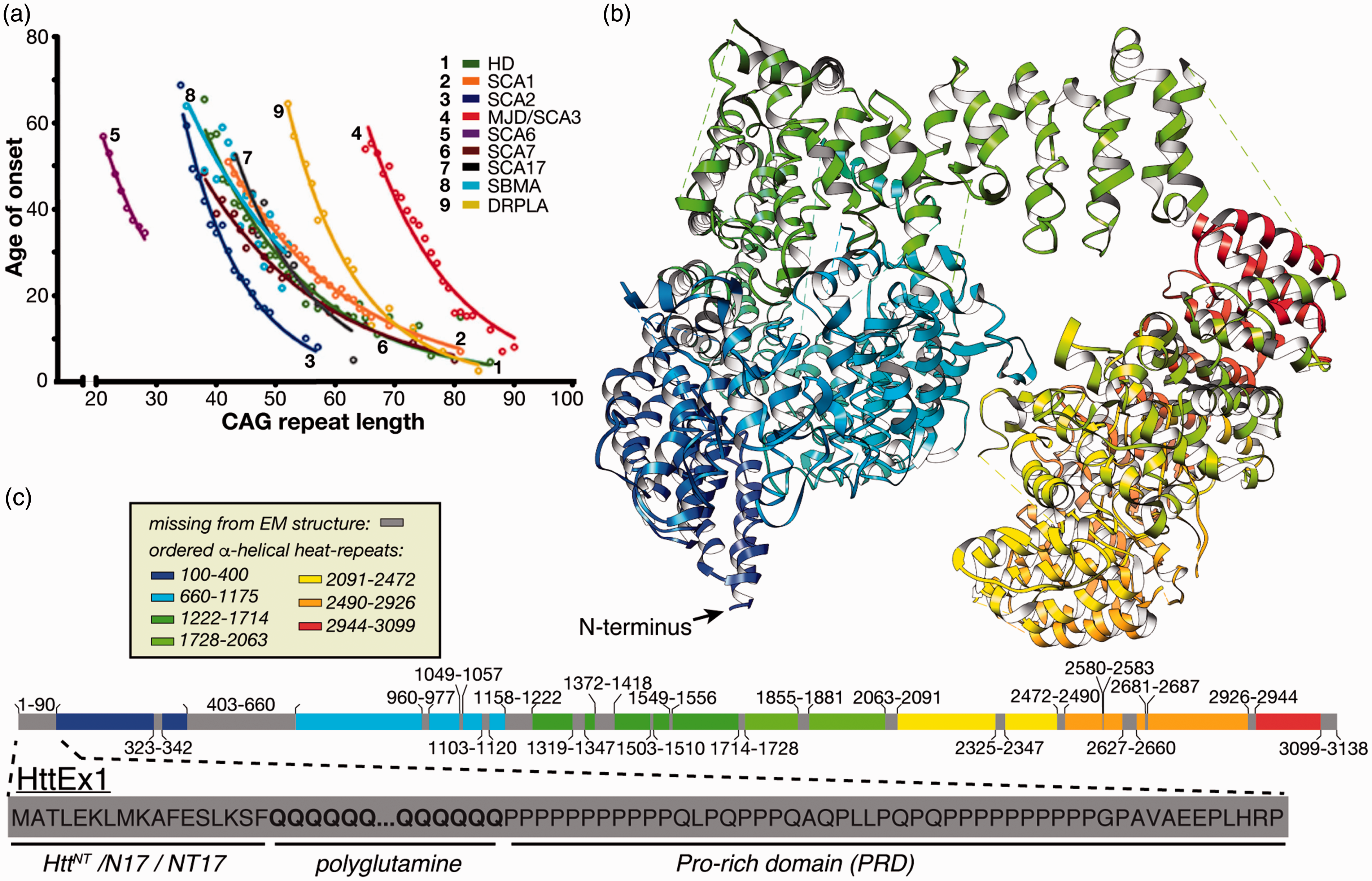
Genetic aspects of HD and other CAG repeat disorders. (a) Age of onset inversely correlates with the extent of expansion, for lengths beyond a disease-specific threshold. The figure was adapted from Kuiper et al. 9 with permission. (b) Wild-type Htt structure solved by cryoEM (PDB 6EZ8; Guo et al. 8 ). (c) Color-coded resolved and invisible domain segments from the Htt cryoEM structure, with the HttEx1 that contains the polyglutamine stretch enlarged below.
HD mutant protein
First documented by George Huntington in 1872, HD causes chorea and cognitive disruptions. 5 It strikes adults and rarely adolescents, but the latter are afflicted with much harsher symptoms. The connection between HD and mutation of the huntingtin (Htt) protein was identified in the 1990s. 6 The age of onset of HD and other polyglutamine expansion diseases is inversely dependent on the length of the CAG repeat (Figure 1(a)). Therefore, HD disease-risk is strongly predicted by its characteristic mutation, in contrast to the genetic and environmental complexities of AD and PD.
Wild-type Htt is a large multidomain protein with functions in various cellular processes. 7 Structurally, Htt is prone to dynamic disorder with the most recognizable folded domains forming so-called HEAT repeats. HEAT repeats are α-helical domains found in a number of proteins, whose names explain the HEAT acronym: huntingtin, elongation factor 3, A subunit of protein phosphatase 2A and TOR1 signaling kinase. The large size of Htt and its inherent disorder presented obstacles to structural determination until 2018 when cryo-electron microscopy (cryoEM) was successfully applied to a complex of full-length wild-type Htt stabilized by huntingtin-associated protein 40 (HAP40), 8 yielding the Htt structure shown in Figure 1(b). Nonetheless, segments constituting almost a quarter of Htt remain invisible in this structure due to high local flexibility, notably including the first exon of Htt (HttEx1) that features the polyglutamine segment mutated in HD (Figure 1(c)).
This inherent disorder of HttEx1 (and its polyglutamine segment) may be functionally relevant as the native role of polyglutamine segments likely depends on their tendency to be flexible and without well-defined secondary structure. Attempts at capturing a polyglutamine domain by X-ray crystallography have yielded structures in which the polyglutamine segment is either invisible or present in a variety of structural states.10–12 Accordingly, polyglutamine domains seem to act as semi-flexible linkers that connect other functionally relevant domains. Polyglutamine’s conformational ensemble may have unique properties necessary for proper positioning of the domains that flank it. 13 Structural studies using fluorescence lifetime imaging microscopy detection of Förster resonance energy transfer (FLIM-FRET) indicate a hinge-like function of polyglutamine domains14,15 showing the importance of intramolecular proximity between N-terminal and proline-rich domains. Others propose an innate ability for interactions with specific protein partners based on a glutamine-rich composition or multi-protein coiled–coil formation.16,17
Aggregation by mutant Htt fragments
The flexible regions of Htt harbor a number of caspase cleavage sites with apparent relevance to HD. 18 Caspase 3 cleavage products formed in vitro are consistent with Htt fragments in HD cerebellum, striatum and cortex, including fragments that map onto HttEx1. 19 A notable, but as yet poorly understood aspect of the different-length Htt fragments is that they cause different levels of toxicity. For instance, in drosophila the HttEx1 fragments exert particularly high toxicity. 20 In human neurons, N-terminal Htt fragments have been identified in neural intranuclear inclusions (NIIs) and dystrophic neurites (DNs) (Figure 2(a)), with their C-terminal counterparts in the cytoplasm.21,22 Remarkably, an HttEx1 fragment also forms via erroneous splicing of the mutant protein. 23
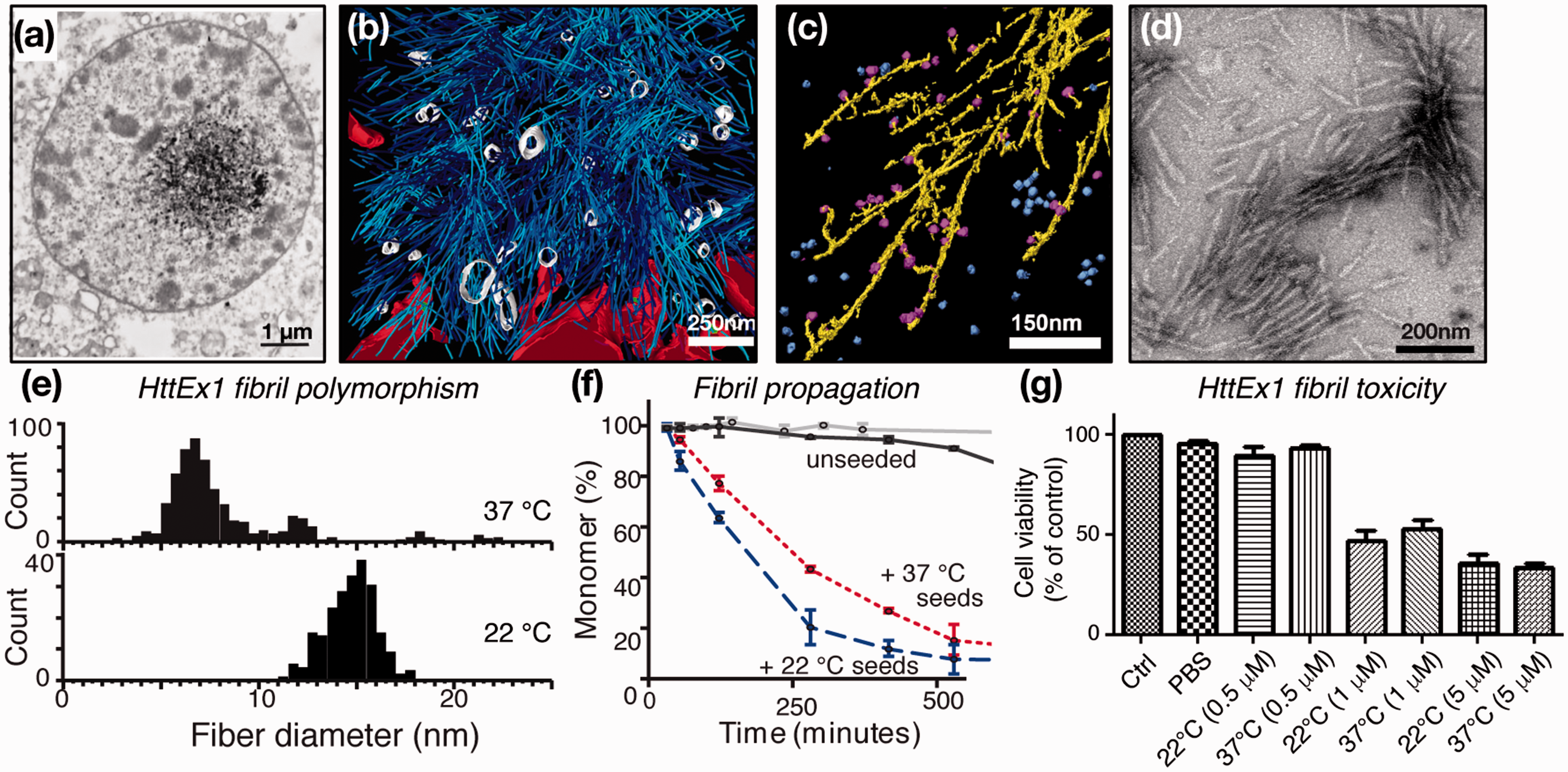
Mutant HttEx1 fibrils in vivo and in vitro. (a) Neuronal inclusions from HD patient; adapted from DiFiglia et al. 21 with permission from AAAS. (b) 3D rendering of tomographic EM showing the interaction zone between an inclusion body and cellular membranes in a HttQ97-transfected HeLa cell. ER membranes (red), ER-bound ribosomes (green), HttQ97 fibrils (cyan), and vesicles inside the IB (white). Adapted from Bauerlein et al. 27 with permission from Cell. (c) Annotated 3D EM tomogram of Q51-HttEx1 fibrils (yellow) in presence of TRiC chaperones (blue/magenta). Adapted with permission from Shahmoradian et al. 64 (d) Negative-stain EM of Q44-HttEx1 fibrils used for ssNMR study. Adapted from Hoop et al. 56 (e) Q44-HttEx1 forms temperature-dependent polymorphs with different widths, which seed polyglutamine protein aggregation (f) and cause neuronal toxicity (g). Panels (e–g) were adapted with permission from Lin et al. 32
The findings described above led to many studies of HttEx1 in model animals and neuronal cells, which commonly observe HD-like symptoms, neuronal degeneration, and HttEx1 inclusions.24–26 A recent cryoEM tomography study provided a view of the structures formed by aggregated HttEx1 in a cellular context. 27 A cluster of filamentous structures is observed (Figure 2(b)), with individual filaments resembling HttEx1 fibrils formed in vitro (Figure 2(c) and (d)). Of potential disease-mechanistic relevance, the fibrils interact with various subcellular organelles, leading, for example, to deformation of the endothelial reticulum (ER) membrane. Thus, μm-sized puncta seen in fluorescence studies likely contain numerous much smaller filaments. In isolation, these filaments are hard or impossible to detect unless super-resolution optical methods are applied.28,29 This distinction may in part underlie the apparent disconnect between observable aggregate load and neurotoxicity, given also that isolated fibrils (assembled in vitro) are toxic to cells but may be small enough to be missed in fluorescence assays (Figure 2(g)).28,30–32
Polyglutamine segments have long been known to undergo self-assembly in vitro. This aggregation propensity depends on the repeat length,33,34 both in polyglutamine peptides and in the context of HttEx1. Morphologically, both polyglutamine peptides and HttEx1 form filamentous assemblies (Figure 2), as seen by negative-stain transmission electron microscopy (TEM) or atomic force microscopy (AFM).27,33,35,36 Crucially, the kinetics of self-assembly are highly dependent on not only the polyglutamine length but also the segments flanking the polyglutamine (Figure 3(a)). 37 The aggregation of HttEx1 is dramatically more efficient than that of the corresponding polyglutamine peptide,38,39 while the flanking regions also affect the aggregates’ morphology and toxicity.37,40,41
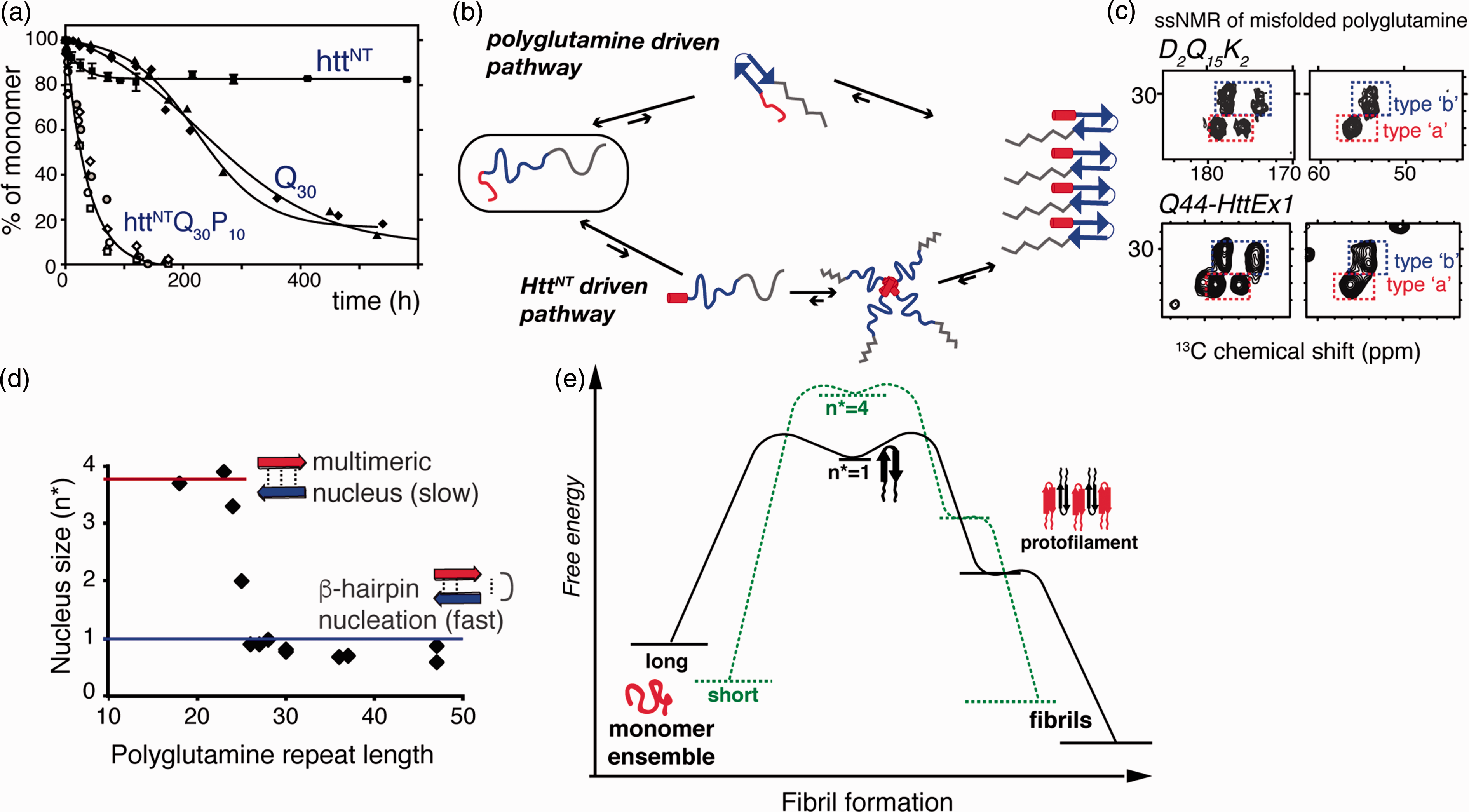
Mechanisms of aggregation. (a) Expanded polyglutamine without (Q30) and with (httNTQ30P10K2) Htt flanking regions attached show dramatically different aggregation kinetics. Adapted from Sivanandam et al. 77 with permission from the American Chemical Society. (b) Intrinsically disordered HttEx1 can aggregate via both polyglutamine driven (top) and HttNT- driven mechanisms (bottom). (c) Both mechanisms yield products with the same ssNMR signals for the polyglutamine fibril core structure (see Figure 4): spectra of Q44-HttEx1 and polyglutamine peptide, D2Q15K2. Adapted from Hoop et al. 56 . (d) Length-dependent nucleus size data96–98 show that fast aggregation of long polyglutamine is facilitated by monomeric nucleation, likely involving β-hairpin formation. (e) Schematic free energy profile of fibril formation and growth, for short and long polyglutamine, based on published nucleation and fiber elongation energy values.96,97,99,100
The enhancement of aggregation has been traced to the N-terminal segment preceding the polyglutamine domain, commonly known as N17, NT17, or HttNT (Figure 1(c)).38,41,42 Note that in vivo the first Met is likely removed to yield a 16-residue HttNT starting with an acetylated Ala. HttNT is critical for the trafficking and localization of Htt and has a highly conserved primary sequence. In isolation, HttNT is in a concentration-dependent equilibrium between disordered monomers and α-helical multimers.38,43,44 Intermolecular interactions, including HttNT self-assembly, stabilize amphipathic α-helical structure in HttNT.10,45,46 The C-terminal proline-rich domain (PRD) of HttEx1 greatly reduces the propensity for aggregation,47,48 but its effect is outweighed by HttNT, when present (Figure 3(a)). PRD-binding proteins, such as FE65, 49 often do so by recognizing the polyproline II (PPII) helical structure of the oligoproline motifs. This same PPII propensity is thought to underpin the aggregation inhibition.38,47,48
Clearly there is an important interplay in the unaggregated protein between the behavior of the polyglutamine domain and its respective flanking domains. Numerous studies, both experimental and computational, have probed this soluble structural ensemble50–52 but in the current review we will focus on structural studies of the misfolded aggregates.
Structural analysis of polyglutamine aggregates
Shortly after the discovery that expanded polyglutamine proteins result in the protein deposition of HD, models of the aggregate structures were proposed. Perutz et al. 53 advocated a structure of pleated antiparallel β-sheets, stabilized by hydrogen bonds between the glutamine side chain and backbone. Early X-ray structural studies of polyglutamine aggregates revealed a cross-β diffraction pattern that is the hallmark of amyloids and amyloid-like structures. 54 Similar data were obtained for both polyglutamine peptides and fibrillar HttEx1, 54 a finding reproduced by later studies.55,56 The fiber diffraction data contain insufficient information to define a unique atomic-level structure but did lead to a new structural hypothesis. First, Perutz et al. 57 proposed a parallel β-sheet-based tubular fold, offering a potential rationale for the HD expansion threshold. However, a later report argued for an alternative explanation of the same data, favoring an antiparallel rather than parallel β-sheet structure, and featuring β-hairpin motifs. 58 Note that the idea of β-hairpin formation during polyglutamine aggregation was invoked very early on by Perutz et al. 54 The antiparallel β-sheet architecture was supported by an independent X-ray study, which reported subtle variations among different-length polyglutamine peptides and proposed distinct structures with kinked side chains. 55 Thus, while X-ray studies provided unambiguous evidence of an amyloid-like architecture that was shared by aggregated polyglutamine and HttEx1, they were unable to resolve a unique atomic structure.
Various other techniques provided important clues regarding the structure of aggregated polyglutamine. Circular dichroism (CD), Fourier-transform infrared spectroscopy (FTIR), and UV resonance Raman (UVRR) spectroscopy indicated the formation of β-sheets, and specifically antiparallel β-sheets.59–63 Notably, the Zanni group combined multidimensional IR with isotopic labeling to observe β-hairpin structures in aggregated K2Q24K2W. 61 As discussed below, β-hairpin motifs were subsequently also detected in the expanded polyglutamine segment of aggregated mutant HttEx1. 32
Recent progress in HttEx1 fibril structure
The recent productive efforts toward a structural understanding of disease-relevant mutant HttEx1 aggregates combined a number of biophysical methods. In isolation, techniques like EM, nuclear magnetic resonance (NMR), and electron spin resonance (ESR) provide incomplete information, but together yield a compelling view of the fibrils’ structure and dynamics. We have already introduced various key contributions made by EM (Figure 2). Moreover, EM and AFM have both revealed other significant characteristics of HttEx1 fibrils, such as their propensity to display branch points that is attributed to surface-mediated secondary-nucleation events.35,64,65 However, unlike recent breakthrough studies of other protein filaments,3,4,66 EM thus far failed to resolve the atomic structure of HttEx1 or polyglutamine aggregates, seemingly due to the fibrils’ structural heterogeneity.27,64,67
NMR studies of fibrillar HttEx1
Liquid-state NMR studies have probed soluble ensembles of polyglutamine-containing peptides and proteins.68–74 This technique excels at providing structural and dynamic data on rapidly tumbling molecules in solution. However, as soon as expanded polyglutamine proteins self-assemble, they quickly form structures that no longer tumble fast enough to be tractable by solution NMR. This is due to the fact that immobilized or slowly tumbling molecules yield very broad spectra with low intensities, thus preventing effective characterization. In recent years, modern solid-state NMR (ssNMR) protocols and instrumentation have made it feasible to study large and immobilized protein assemblies, even in presence of dynamic and static disorder,75,76 resulting in ssNMR becoming an essential tool for amyloid protein research.2,75 Using magic angle spinning (MAS), the immobilized large protein assemblies are rapidly rotated in the magnetic field to generate high-resolution ssNMR spectra, independent of assembly size. The initial applications of ssNMR to polyglutamine-based aggregates made less than a decade ago71,77 immediately revealed a number of intriguing and important features. First, despite dramatic differences in aggregation kinetics or propensity, different labs consistently reported a highly characteristic “signature” for the self-assembled misfolded state of the polyglutamine stretch itself (Figures 3(c) and 4(a)). This ssNMR signature consistently combines highly atypical resonance frequencies with two equally populated sets of distinct NMR signals, in a unique combination that is only seen in polyglutamine aggregates and not in any other proteins studied by solution or solid-state NMR. 78 The latter peak doubling arises even when a single residue is labeled, indicating a structural heterogeneity on the single-residue level.
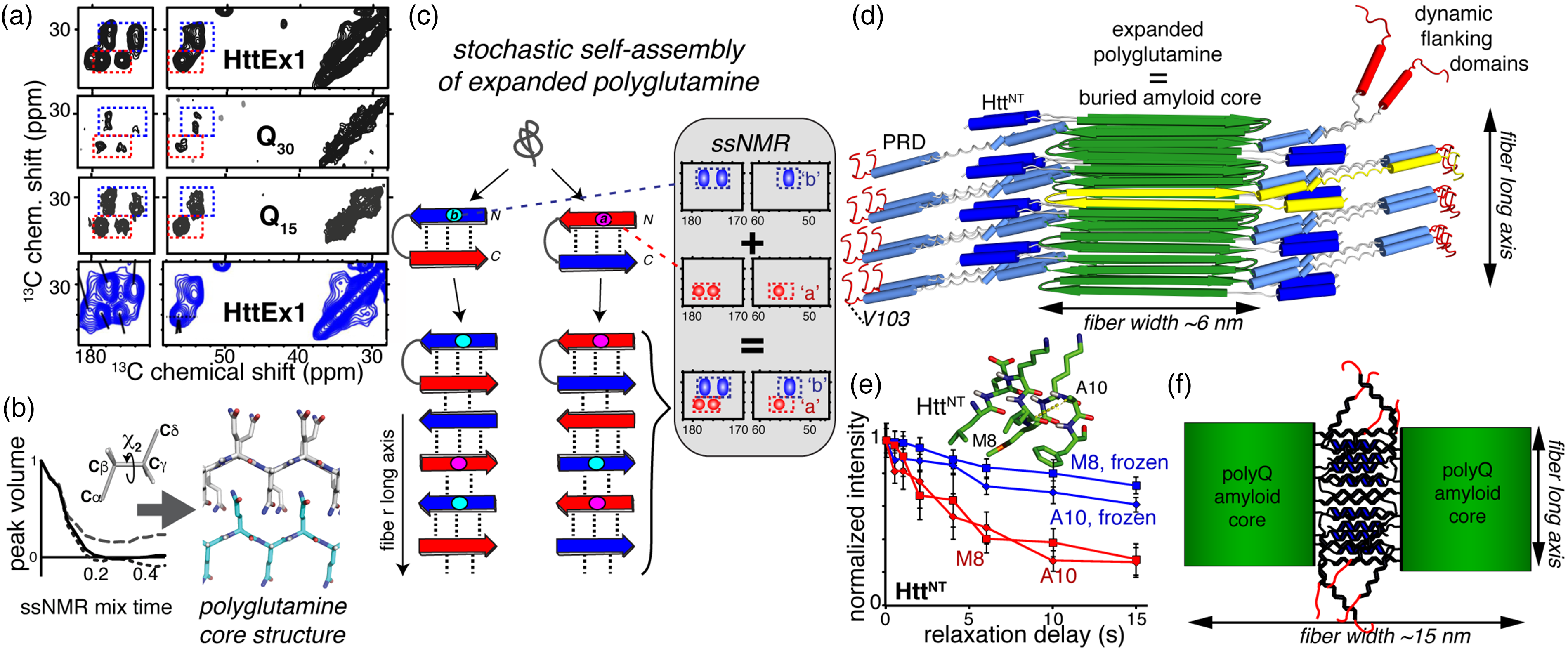
SSNMR structural studies of misfolded fibrils. (a) Fibrillar polyglutamine gives an identical ssNMR signature, whether in Q44-HttEx1 fibrils (top), Q46-HttEx1 fibrils (bottom), or polyglutamine peptides with 15 or 30 glutamines. Data from Lin et al. 32 and Sivanandam et al. 77 Data for Q46-HttEx1 were adapted with permission from Isas et al. 79 copyright 2015 American Chemical Society. (b) Tailored ssNMR dihedral angle measurements probe the polyglutamine core structure, and reveal two differently structured β-strand types (adapted from Hoop et al. 56 ). (c) A stochastic assembly process of the alternating β-strand structures explains the ssNMR spectral signature of polyglutamine amyloid. Adapted with permission from Hoop et al. 56 (d) Architecture of mutant HttEx1 fibrils derived from ssNMR and EM constraints. (e) SSNMR relaxation measurements show dynamic changes in the solvent-exposed HttNT α-helical segment upon solvent freezing. Adapted from Hoop et al., 80 with permission from the American Chemical Society. (f) Schematic illustration of flanking-domain interactions enabling higher order assembly of the wider HttEx1 fibril polymorphs shown in Figure 2(e). Panels (d) and (f) are adapted with permission from Lin et al. 32 (A color version of this figure is available in the online journal.)
Structural ssNMR measurements and mechanistic studies provide a compelling rationale for this surprising, but reproducible, doubled ssNMR signature (see below; Figures 3(c) and 4(a)). First, ssNMR measurements of backbone and side chain torsion angles confirmed that the two signals derive from two distinct conformations (Figure 4(b)). 56 Inter-residue ssNMR correlations show that each of these conformers is present in surprisingly long, uninterrupted stretches.56,74,77–80 More specifically, they form equal amounts of two distinct types of uninterrupted β-strands. The two strands differ in their side chain torsion angles, but within each strand all residues have the same backbone and side chain geometry (Figure 4(b)). Both β-strands do share a 180° χ2 angle – implying an extended side chain structure 56 akin to the steric zipper concept seen in other amyloids. 81 A recent ssNMR study that employed dynamic nuclear polarization (DNP) allowed for the structural fingerprinting of unlabeled HttEx1 aggregates and provided further evidence for their antiparallel β-sheet assembly.56,82 In such an antiparallel arrangement, the two ssNMR-revealed β-strand types appear capable of inter-strand hydrogen bonding to each other (but not themselves). An assembled antiparallel β-sheet requires the presence of equal amounts of the two complementary β-strand geometries. 83 Thereby, these findings provide a rationale for the polyglutamine amyloid ssNMR signature featuring equal amounts of the two corresponding sets of ssNMR signals. All ssNMR studies of polyglutamine-based aggregates with a single labeled residue show both signals for that labeled residue.56,77,78,80,84 This points to a stochastic assembly process in which any glutamine segment (or residue) has a 50/50 chance of adopting either of the two complementary β-strand motifs (Figure 4(c) 56 ).
Polyglutamine length and the HD disease threshold
Polyglutamine retains solubility up to a length of approximately seven residues,44,85 but peptides of eight or more (well below the HD threshold) are routinely studied in their aggregated state. Interestingly, polyglutamine peptides with 15 to over 50 residues yield identical chemical shift patterns by ssNMR that are indistinguishable from those of HttEx1 fibrils’ amyloid core (Figures 3(c) and 4(a)).71,77,79,80,86 The ssNMR chemical shifts are very sensitive to changes in structure and typically show clear differences between amyloid polymorphs with different core structures. 87 In ab initio calculations, the experimental ssNMR chemical shifts are inconsistent with the pre-ssNMR structural models of polyglutamine discussed above. 56 Therefore, these ssNMR data are in apparent contradiction with reports arguing for dramatic changes in architecture between short and long polyglutamine aggregates.55,88,89
Does this imply there is no length-dependent transition in the misfolded structure or misfolding mechanism as a rationale for the disease threshold? One potential rationale has been proposed that integrates much of the available structural and mechanistic data. Mixed-isotope ssNMR experiments, reminiscent of the above-mentioned IR study, 61 identified intramolecularly hydrogen-bonded β-hairpins in the amyloid core of Q44-HttEx1 fibrils (Figure 4(c) and (d)). 56 The weak intensity from non-β-strand residues indicated that no more than a single turn was present. This implied 20-residue strands constitute the β-hairpin, which is reminiscent of the detection in a prior ssNMR study of surprisingly long strand segments in polyglutamine aggregates. 71 Their study of a 15-residue polyglutamine peptide with isotopic labels mid-peptide did not reveal any turn structure signals. The Q15 peptides featured a single β-strand per peptide, while the β-hairpin Q44-HttEx1 fibrils contained two 20-residue strands (Figure 4(d)). Thus, the propensity for β-hairpin formation during polyglutamine (or HttEx1) aggregation would depend on the polyglutamine expansion length, with direct implications for the aggregation kinetics. In this model, β-hairpin formation may be required to achieve a sufficiently high aggregation propensity that cannot be suppressed by the cellular protein homeostasis machinery (under in vivo conditions). Interestingly, the currently available in vitro data (Figure 3(d)) point to the switch to monomeric nucleation occurring well below the typical disease thresholds. This could imply that β-hairpin-mediated aggregation is necessary but not sufficient for disease onset. Alternatively, there may be an as-yet unknown effect of environmental (cellular) factors modulating the polyglutamine aggregation process in vivo, dictating whether or not a particular polyglutamine length follows a β-hairpin-driven aggregation process or not.
HttEx1 polymorphism
Various studies have noted that a particular HttEx1 protein can adopt multiple types of misfolded or aggregated structures (e.g. Figure 2(e)).31,60 Such amyloid polymorphism is also common in other disease-associated protein aggregates. 87 Early studies suggested that HttEx1 polymorphism stems from an architectural change of the polyglutamine stretch. 60 However, ssNMR experiments consistently report identical signatures for the polyglutamine stretch, with polymorph-dependent differences localized to the non-amyloid flanking segments. This suggests that expanded polyglutamine forms a reproducible protofilament structure that can co-assemble into different supramolecular architectures, stabilized by variable flanking region interactions. 32 Conceptually analogous “supramolecular polymorphs” 3 have subsequently been described in several other disease-related amyloids, indicating that this may be a more general phenomenon.3,4,66,90,91
Both ssNMR and ESR studies have been instrumental in probing the fate of the flanking regions in HttEx1 post-aggregation.32,45,74,77,79,80,84,86 In contrast with the rigid and well-structured polyglutamine motif, both flanking segments are consistently found to display significant dynamics. The greatest flexibility is associated with the C-terminal end of HttEx1, which usually retains similar dynamics and accessibility before and after aggregation.32,79,92,93 Approaching the rigid polyglutamine amyloid core, the PRD becomes increasingly rigid, presumably due to intermolecular interactions.32,79,92,93 The HttNT flanking segment also has increased mobility and solvent accessibility (Figure 4(d) and (e)) relative to the polyglutamine fibril core.32,77,79,80 The reported dynamics and secondary structure of the HttNT varied among different studies, which may relate to a combination of fibril polymorphism, differences in protein constructs employed, and different fibril formation protocols. In our own work,32,77,84 ssNMR studies of HttNT in the fibrils consistently point to an α-helical conformation and a partial, though incomplete, immobilization (Figure 4(e)), likely facilitated by dense packing of the flanking segments on the fibril surface (Figure 4(d)).
When comparing different HttEx1 polymorphs (Figure 2(e)), ssNMR studies show clear differences in the dynamics and solvent accessibility of the flanking segments.32,93 Antibodies that recognize specific parts of the HttEx1 sequence also bind differently. 32 The variable flanking segment exposure in HttEx1 polymorphs points to a mechanism for stabilizing the supramolecular polymorphs (Figure 4(f)), leading to the distinct fiber widths observed by EM (Figure 2(e)). These differences in burial or exposure of the flanking sequences should also modulate the biological (e.g. toxic) activity of the misfolded protein. For example, exposed and dynamic79,80,92 HttNT are implicated in membrane interactions, and their accessibility will affect the filaments’ ability to bind cellular membranes.27,94,95 The flanking segments’ varying ability to engage with biological membranes, chaperones, and other protein binding partners will have direct relevance for various pathogenic mechanisms.
Biological and mechanistic implications
The increased availability of structural information has greatly improved our understanding of the mechanisms by which polyglutamine proteins misfold, aggregate, and contribute to disease. Like other amyloidogenic processes, polyglutamine aggregation is nucleation driven. The nucleation event is the rate-limiting step with a positive free energy (ΔG) and very small equilibrium constant for nucleus formation (Kn*) 96 (Figure 3(e)). Once a minimal nucleus is formed, the elongation process is thermodynamically favorable and spontaneous.99,101 For polyglutamine, Kn* increases with longer repeat lengths, leading to ever faster aggregation of proteins with longer polyglutamine repeats. 96 Thus, Htt fragments with very long glutamine repeats nucleate more quickly than those with short repeats. Why does Kn* show this length dependence, and which conformation can lower the free energy of nuclei for longer repeat lengths? The size of the critical nucleus depends on polyglutamine length (Figure 3(d))100,101: although short polyglutamines aggregate, they require a multimeric nucleation event. The rate-limiting step for long polyglutamine manifests instead as a monomeric event.100,101 Various lines of evidence support the idea that monomeric nucleation reflects the formation of a β-hairpin within the expanded polyglutamine monomer. Structural studies detect β-hairpin structures in the end-product of aggregated polyglutamine and Q44-HttEx1.56,61 Moreover, β-hairpin-favoring mutations accelerate aggregation kinetics and increase fiber stabilities, without changing the characteristic polyglutamine ssNMR signature. 78
Molecular dynamics (MD) simulation studies can examine transient events and dynamic processes that are hard to probe experimentally. Given the focus on this review, we refer readers to other recent articles.50,51 We do note an interesting set of MD studies that probed polyglutamine folds that may be compatible with the noted “monomeric” mechanism,100,102 since they favored β-hairpin motifs over less stable β-nanotube or β-pseudohelix conformations.
As noted above, all polyglutamine aggregate studied by ssNMR to date share a common characteristic ssNMR signature. One intriguing possibility is that this reflects a common core structure that may extend to all polyglutamine proteins associated with disease and that all the expanded-polyglutamine proteins aggregate via an analogous misfolding pathway. The different disease thresholds may relate to the impact of protein context (i.e. flanking domains) on the propensity for the polyglutamine region to adopt an elongation-capable β-hairpin. In HD, the flanking regions are dynamically disordered, allowing the polyglutamine segment termini to readily approach each other. Yet, in other polyglutamine proteins, combinations of steric interactions and/or electrostatic repulsion of the flanking domains may hinder β-hairpin formation. On the other hand, the protein fold of the α1 subunit of the neuronal P/Q-type voltage-gated calcium channel (associated with SCA6 disease103,104) may place its polyglutamine stretch in a conformation that encourages β-hairpin formation. This could rationalize the reduced threshold seen in SCA6, compared to the other polyglutamine expansion diseases 104 (Figure 1(a)). Further structural and mechanistic studies are needed to test these proposals.
Oligomerization
Some reports claim that long polyglutamine peptides form small spherical oligomers (dimers and trimers) that coexist with larger ones in solution. 89 However, other studies offer compelling evidence that polyglutamine de novo aggregation proceeds without formation of defined oligomers.96,97 There is more evidence for the formation of prefibrillar oligomeric assemblies for expanded HttEx1.105–108 In cells, wild-type HttEx1 can undergo a type of liquid–liquid phase separation, while polyglutamine-expanded HttEx1 assembles into more compact irreversible inclusions. 109 When studied structurally, HttEx1 oligomers generally appear to be α-helical, rather than β-sheet-rich like Aβ oligomers.38,44,77,110 This helical structure is stabilized by bundling of α-helical HttNT, with the α-helical structure likely extending into the initial parts of the polyglutamine.44,77
The above differs notably from the extensively studied oligomers formed by the AD Aβ peptide. Aβ oligomers feature antiparallel β-sheet structures, which are distinct from the in-register parallel β-sheet fibrils, and which by many are proposed to reflect β-hairpins. An intriguing possibility is that β-hairpin formation may similarly occur during polyglutamine and Aβ aggregation, but that (unlike polyglutamine) the Aβ β-hairpin motif is not accommodated in the filamentous structure, instead becoming trapped in the semi-stable oligomeric intermediates.
The role of aggregates in disease
There is an ongoing debate whether Htt-related aggregates in the brain are cytotoxic and cause disease development, or the toxicity comes from other species, such as soluble oligomers or even misfolded monomers. Some studies showed no correlation between Htt aggregates and cell toxicity.111,112 On the other hand, many studies have shown toxic effects of HttEx1 or polyglutamine aggregates on neuronal cells.60,113–115 Various rationales have been proposed to unify these seemingly contradictory findings, and we will discuss some with an eye on the current knowledge of HttEx1 aggregate structure. First, as noted above, fluorescence-based assays commonly used in evaluating aggregate load may not reliably measure sub-diffraction sized protein deposits. As illustrated in Figure 2, individual filaments have widths on the nm-scale and are thus orders of magnitude smaller than visible inclusions. Thus, cells lacking μm-sized puncta may nonetheless contain significant amounts of protein filaments. Second, different polymorphs can exhibit substantially different levels of toxicity. The toxicity of polyglutamine proteins is dependent of the flanking sequences, protein–protein interactions, and fibril polymorphism.40,60 For example, in a mouse that expressed a truncated fragment of expanded Htt that is longer than HttEx1 (shortstop mouse), the existence of neural inclusions did not lead to neural abnormalities or degeneration. 112
This also raises the fundamental question of the mechanism of toxicity. In our view, this issue remains largely unresolved. One mechanism specific to polyglutamine proteins is the recruitment or sequestration of other proteins featuring polyglutamine repeats by the aggregates, which similarly affects wild-type and expanded polyglutamine proteins. Thus, the cellular concentrations of essential proteins could be lowered to the point of dysfunction or toxicity. A study that attempted to test this mechanism using D-amino-acid polyglutamine aggregates found that even these fibrils induced toxicity in PC12 neuronal cells, but also offered a potential structural rationale by which observed recruitment could cross the chiral barrier. 115 Aggregated Htt is also thought to impair intracellular trafficking into organelles such as the mitochondria and nucleus, potentially by interacting with the import/export protein machinery. Moreover, polyglutamine protein inclusions sequester many non-polyglutamine proteins, including components of the protein quality control machinery. Thus, fibrils are known to have various detrimental effects. There is also a growing interest in the finding that Htt fibers are able to propagate from one cell to another, enabling disease propagation via a prion-like process.116,117 This phenomenon requires the fibers to have conformations amenable to them crossing cellular membranes and seeding the self-assembly of other proteins in nearby cells. In other words, the polymorphism of Htt-derived aggregates, which we are only just beginning to understand, can likely dramatically alter their biological behavior and pathogenic effects in a way that is orthogonal to the apparent aggregate load.
Counteracting and controlling protein aggregation
Nature has deployed cellular protection mechanisms to counteract disease-causing protein misfolding and aggregation. Molecular chaperones refold misfolded proteins and prevent pathogenic protein aggregation.118–120 Chaperones such as TRiC, DNAJB6 and DNAJB8 have been shown to inhibit Htt aggregation.64,121,122 The proposed molecular mechanisms by which these chaperones act are mirrored in the abovementioned structural data for the aggregates. TRiC binds the α-helical HttNT that was detected by ssNMR in the HttEx1 fibrils, while the mentioned DNAJ co-chaperones appear to inhibit primary nucleation by recognizing β-hairpins. Thus, the fiber structures mirror the very structural features that can be targeted for the inhibition or modulation of aggregation. Along similar lines, post-translational modifications (PTMs) offer a mechanism for modulating disease risk and aggregation. Phosphorylation of HttNT is dependent on the glutamine repeat length123, 9 and changes aggregation and toxicity.84,125 The clustering of the PTMs’ repulsive charges in the densely packed HttNT in misfolded Htt (Figure 4) leads to a destabilization that enhances the neurons’ ability to target and clear the Htt aggregates. Thus, insights into the structures of disease-associated protein deposits can direct efforts to design novel or enhanced treatment strategies. One critical component in such efforts will be the delineation of the structural variables (e.g. supramolecular architecture and/or exposure of flanking domains) that most strongly predict biological functions such as neuronal toxicity, membrane interactions, and inter-neuron propagation. We hope that progress made in understanding HD may also inform our understanding (and treatment) of more complex NDDs like AD, PD and ALS.
Footnotes
Authors’ contributions
Both authors performed the literature review, preparation of figures, writing and reviewing of the manuscript.
ACKNOWLEDGMENTS
We thank James Conway, Matt Lee, Mingyue Li, and Talia Piretra for their careful reading of the manuscript and their insightful suggestions.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We acknowledge funding from NIH grants R01 GM112678 and AG019322 supporting our polyglutamine research.