
Abstract
Objective. Ethanolic extract of Gymnema sylvestre (GS) leaves is used as a potent antidiabetic drug in various systems of alternative medicine, including homeopathy. The present study was aimed at examining if GS also had anticancer potentials, and if it had, to elucidate its possible mechanism of action. Methods. We initially tested possible anticancer potential of GS on A375 cells (human skin melanoma) through MTT assay and determined cytotoxicity levels in A375 and normal liver cells; we then thoroughly studied its apoptotic effects on A375 cells through protocols such as Hoechst 33258, H2DCFDA, and rhodamine 123 staining and conducted ELISA for cytochrome c, caspase 3, and PARP activity levels; we determined the mRNA level expression of cytochrome c, caspase 3, Bcl2, Bax, PARP, ICAD, and EGFR signaling genes through semiquantitative reverse transcriptase polymerase chain reaction and conducted Western blot analysis of caspase 3 and PARP. We also analyzed cell cycle events, determined reactive oxygen species accumulation, measured annexin V-FITC/PI and rhodamine 123 intensity by flow cytometry. Results. Compared with both normal liver cells and drug-untreated A375, the mortality of GS-treated A375 cells increased in a dose-dependent manner. Additionally, GS induced nuclear DNA fragmentation and showed an increased level of mRNA expression of apoptotic signal related genes cytochrome c, caspase 3, PARP, Bax, and reduced expression level of ICAD, EGFR, and the anti-apoptotic gene Bcl2. Conclusion. Overall results indicate GS to have significant anticancer effect on A375 cells apart from its reported antidiabetic effect, indicating possibility of its palliative use in patients with symptoms of both the diseases.
Introduction
The burden of cancer is increasing worldwide despite advances made in diagnosis and treatment with the orthodox system of medicine. Skin cancer is the third most common among the human malignancies 1 arising from the pigmented cells of the skin called melanocytes. Epidemiological studies have shown that many types of cancers may be avoidable,2,3 and in many, chemotherapy and radiations are highly ineffective necessitating other options for the cure of this disease. 4 Evidence-based studies reported that melanoma is highly resistant to conventional treatments such as radiation and chemotherapy. 5 Complementary and alternative medicine is increasingly becoming popular particularly in oncology, which often applies various complementary and alternative medicines to give the patients a better quality of living by alleviating their sufferings.6,7
In traditional systems, medicinal plants and plant-derived drugs are extensively used, but many of them need scientific validation to give confidence to the users, particularly when treating chronic life-threatening conditions such as cancer.8,9 Gymnema sylvestre is a woody climber tree, commonly known as Gurmar in India, belonging to the order Gentianales, and family Asclepiadaceae. The plant grows in the western and central parts of India, in tropical Africa, and in Australia. Its ethanolic extract is considered a potent antidiabetic drug used in folk medicine, Ayurveda, and homeopathic systems of medicine, 10 but whether it has anticancer property has never been tested earlier. Hence, as a part of our routine program of testing extracts of some unconventional and unexplored plants for their possible anticancer potentials, we undertook the present study. We tested the ethanolic extract of G sylvestre on a human melanoma skin cancer cell line A375, with a view to examining if a drug with dual protective role against diabetes and cancer could be added in the repertory for facilitating newer drug design.
Materials and Methods
Reagents
Dulbecco’s modified Eagle medium, fetal bovine serum, penicillin, streptomycin, neomycin antibiotic, trypsin, and ethylenediamine tetraacetic acid were purchased from Gibco BRL (Grand Island, NY). Tissue culture plastic wares were obtained from BD Biosciences (San Jose, CA). All organic solvents used were of high-performance liquid chromatography grade. Hoechst 33258, annexin V-FITC, propidium iodide (PI), and MTT [3-(4, 5-dimethyl-thiazol-2-yl)-2, S-diphenyl tetrazolium bromide] were purchased from Sigma Aldrich Co (St Louis, MO).
Cell Line and Cell Culture
Human skin melanoma cell line (A375) and normal liver cell line (WRL-68) were collected from National Centre for Cell Science, Pune, India. Both types of cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and 1% antibiotic (penicillin, streptomycin, neomycin) at 37°C in a humidified incubator (Thermo Electron Corporation, Waltham, MA) with 5% CO2.
Drug
Ethanolic extract of leaves of G sylvestre (GS; homeopathic mother tincture) was purchased from HAPCO (Kolkata, India). Total extract were evaporated to dryness at <50°C. The extract was further dissolved in 70% ethanol to prepare the stock solution for further use in experiments.
Cell Viability Assay
Cell viability of A375 and WRL-68 cells was estimated by the MTT assay 11 as described previously with some modifications. Briefly, cells were dispensed in 96-well flat-bottom microtiter plates at a density of 3 × 102 cells well. After 24-hour incubation, A375 cells were treated with different concentrations (0-300 µg/mL) of GS for 12 and 24 hours, respectively, whereas cell viability assay of WRL-68 cells was done with different concentrations (0-980 µg/mL,) for 24 hours of treatment only.
Detection of Intracellular Reactive Oxygen Species Accumulation and Mitochondrial Membrane Potential (ΔΨm) Determination
The levels of reactive oxygen species (ROS) and ΔΨm were examined by flow cytometry using H2DCFDA and rhodamine 123, respectively, according to methods described previously.12,13 After treatments with GS, cells were loaded with 20 µM H2DCFDA for ROS measurement and 10 µM rhodamine 123 for ΔΨm determination incubated at 37°C for 30 minutes in the dark. Cells were then collected, washed, and resuspended in phosphate-buffered saline (PBS) and analyzed immediately using flow cytometry.
Assessment of Chromatin Condensation
To determine the apoptotic cells after 24 hours of treatment, they were separately stained with Hoechst 33258 (1 µg/mL). Then the cells were analyzed under fluorescence microscope (Axiscope plus 2, Zeiss, Jena, Germany) and representative photographs were taken for further quantitative analysis.
DNA Fragmentation Assay
A total of 1 × 106 cells were treated with various concentrations of GS for 24 hours and then collected by centrifugation. DNA lysis buffer was used to lyse the cell pellets and were incubated overnight with proteinase K (0.1 mg/mL). DNA extraction was done by phenol–chloroform (1:1) method. The DNA was separated in 2% agarose gel, which contains ethidium bromide and visualized by UV trans illuminator.
Annexin V-FITC/PI Assay
Annexin V/PI was used to detect early and late apoptotic cells during apoptotic progression. After treatment with GS for 24 hours the cells were trypsinized, washed twice with cold PBS, and then adjusted to 5 × 105 cells/500 µL in binding buffer containing annexin V-FITC (1 µg/mL) and PI, and finally analyzed by flow cytometry. 14
Analysis of Apoptosis by Flow Cytometry Using TUNEL Assay
DNA strand break analysis was performed by labeling with Br-dUTP following the method of Darzynkiewicz et al. 15 Briefly, 1 × 105 to 2 × 105 cells were suspended in 0.5 mL PBS. This suspension was transferred into a 5-mL polypropylene tube containing 4.5 mL of ice-cold 1% formaldehyde in PBS. Cell pellet was resuspended in 10 mL of FITC-conjugated anti-BrdU mouse antibody solution (1:1000) procured from Abcam (Cambridge, MA). Cells were incubated at room temperature for 1 hour. One milliliter of PI staining solution was added. Cells were incubated for 30 minutes at room temperature and for 20 minutes at 37°C in the dark; after incubation, the fluorescence emissions were analyzed with a flow cytometer (FACS Calibur, BD Biosciences, San Jose, CA).
Preparation of Cell Extract
A total of 1 × 106 cells were lysed in 20 µL of ice-cold lysis buffer(10 mM Tris–HCl, pH 7.5, 1 mM MgCl2, 1 mM ethylenediamine tetraacetic acid, 0.1 mM PMSF, 5 mM β-mercaptoethanol, 0.5% CHAPS, 10% glycerol). Cells were incubated for 30 minutes on ice and centrifuged for 20 minutes at 8000 × g at 4°C. After centrifugation, supernatants were collected and stored at −20°C for further use.
Changes in Activity of Different Apoptotic Signal Proteins
Activity of pro-apoptotic proteins was assessed by indirect ELISA as described earlier. 16 The activities of cytochrome c, caspase-3, and poly(ADP-ribose) polymerase (PARP) were measured with A375 cell lysate, using an ELISA reader (Thermo Scientific, Waltham, MA). All the primary and secondary antibodies were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). pNPP was used as an color developing agent and the color intensity measured at 405 nm wavelength.
RNA Extraction and Semiquantitative RT-PCR Analysis
Total RNA was extracted from the A375 cells using Trizol reagent according to the method described earlier. 17 Fluorescence intensity of band on the agarose gel was measured by using the “image J” software.
Immunoblotting
A total of 50 µg of cell lysate was used for SDS-PAGE (12.5%) electrophoresis to estimate PARP and caspase 3 activities; the cell lysate was then transferred to polyvinylidene difluoride membrane. After blocking with 3% bovine serum albumin, the membrane with specific primary antibodies was incubated overnight at 4°C. The membrane was further incubated for 2 hours with alkaline phosphatase–conjugated secondary antibody. BCIP-NBT was used as developer and the band intensities were measured densitometrically using image J software.
Statistical Analysis
Data were analyzed and significance of the differences between the mean values was determined by one-way analysis of variance with Dunnett’s post hoc tests, using SPSS 14 software. Statistical significance was considered at P < .05.
Result
Effect of GS on Cell Viability
MTT assay showed that GS had significantly cytotoxic effects on A375 cells whereas the normal liver cells (WRL-68) showed no such significant cytotoxicity in vitro (Figure 1B).Treatment of A375 cells with 150 µg/mL (D1), 200 µg/mL (D2), and 250 µg/mL (D3) for 24 hours resulted in approximately 55.41%, 50.64%, and 45.1% cell viability, respectively (Figure 1A). We selected these 3 doses of drugs for our experimental purposes.

(A) Effect of Gymnema sylvestre (GS) on cell viability in skin cancer cell. Cells were cultured in growth media containing different concentrations of GS for 12 and 24 hours. (B) Effect of GS on cell viability in normal liver cell (WRL-68). Cells were cultured in growth media containing different concentrations of GS for 24 hours.
Effect of GS on ROS Generation and Mitochondrial Membrane Potential (ΔΨm) Depolarization in A375 Cell
We observed a time-dependent increase of ROS production and highest percentage of ROS was observed at 3 hours (Figure 2 A-H) of treatment with GS. We found that the rhodamine 123 fluorescence intensity was reduced, thereby reflecting the loss of ΔΨm, which was also confirmed by FACS analysis, showing a marked decrease in fluorescence intensity after of GS treatment (Figure 2 I-P).

Gymnema sylvestre (GS) treatment increased cellular reactive oxygen species (ROS) accumulation (A-H) in A375 cells. Cells were untreated or treated with 200 µg/mL for 1, 2, and 3 hours, respectively, and then incubated with H2DCFDA (20 µM) for 30 minutes prior to fluorescence microscopic and flow cytometric analysis. (A, B) Control cells. (C, D) GS treated for 1 hour. (E, F): GS treated for 2 hours. (G, H) GS treated for 3 hours. The effects of GS on reduction of ΔΨm (I-P) in A375 cells. Cells were either untreated or treated with 200 µg/mL (D2) of GS for 12, 18, and 24 hours, respectively, and were stained with rhodamine 123. After staining, the cells were viewed by fluorescence microscope. (I, J) Control cells. (K, L) GS treated for 12 hours. (M, N) GS treated for 18 hours. (O, P) GS treated for 24 hours.
Effect of GS on Chromatin Condensation
The untreated A375 cells did not take positive staining with Hoechst 33258 (Figure 3A) and showed no cells with visible chromatin condensation. However, with different concentrations of treatment, cells with chromatin condensation appeared to increase in number along with the increase in dose (Figure 3B-D).

Hoechst 33258 staining (A-D) and DNA fragmentation assay (E). After treating with Gymnema sylvestre (GS) for 24 hours, cells were stained with Hoechst 33258 fluorescent dye. More brightly stained cells represent greater quantity of chromatin condensation in the cells. In (E): Ln 1, control cells; Ln2, 150 µg/mL (D1) drug-treated cells; Ln3, 200 µg/mL (D2) drug-treated cells; Ln4, 250 µg/mL (D3) drug-treated cells.
Increased DNA Fragmentation due to Drug Treatment
We examined the DNA fragmentation by agarose gel electrophoresis. Figure 3E indicates a significant increase in internucleosomal DNA fragmentation of A375 cells. When the DNA isolated from GS-treated cells was subjected to agarose gel electrophoresis, a DNA smear characteristic of DNA breakage was observed in the cells treated with different concentrations of GS.
Changes in Percentages of Apoptotic Cells
The data obtained from flow cytometric analysis after staining with annexin V-FITC are summarized in Figure 4A-D. The percentages of apoptosis in GS-treated (150 µg/mL, 200 µg/mL, 250 µg/mL for 24 hours) population were found to be 23.42%, 25.37%, and 31.89% for A375 cells, respectively, as compared with control cells (1.77%).

Flow cytometric analysis. Cells were analyzed after incubation with Gymnema sylvestre (GS) for 24 hours by flow cytometry for annexin V-FITC/PI (A-D) and TUNEL assay (E-H).
GS-Induced Change in TUNEL Positive Cells
GS-induced apoptotic signals were also detected by TUNEL assay in FACS. It was observed that a significant number of TUNEL positive cells were present in the drug-treated cells (Figure 4E-H).
Modulation of Apoptotic Signal Proteins
Cytochrome c, caspase 3, and PARP showed a higher level of expression in contrast to the untreated control group as revealed from indirect ELISA assay. Expression level in GS-treated groups increased in a dose-dependent manner (Figure 5).

Analysis on protein level expression by ELISA. After incubation for 24 hours with Gymnema sylvestre (GS) at 150 µg/mL, 200 µg/mL, and 250 µg/mL, expression of cytochrome c, caspase 3, and PARP were detected by ELISA.
Western blot data also suggested the upregulation of caspase 3 in a dose-dependent manner. Cleavage of PARP was observed on treatment of the drug (Figure 6).
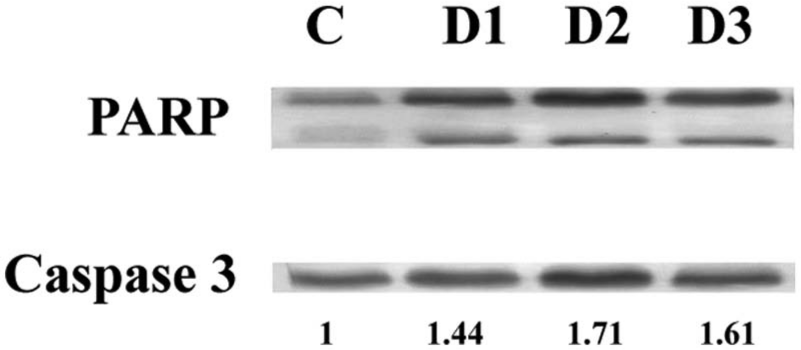
Western blot analysis of PARP and caspase 3. The expression of PARP and caspase 3 was measured in A375 cells on drug treatment. Drug doses were as follows: D1 = 150 µg/mL, D2 = 200 µg/mL, and D3 = 250 µg/mL. The expression of caspase 3 was upregulated and cleaved PARP has been observed.
Changes in mRNA Level Expression
Results of RT-PCR confirmed that there was a significant difference of mRNA expression in between the control and drug-treated groups. After drug treatment, mRNA expression of cytochrome c, caspase 3, Bax, and PARP (Fi increased whereas expression level decreased in case of Bcl2, ICAD (inhibitor of caspase-activated DNase), and EGFR (epidermal growth factor receptor; Figure 7A). Fluorescence intensity of PCR bands on the agarose gel was analyzed by “image J” software (Figure 7B). The primer sequences of amplified genes are given in Table 1.

Analysis of mRNA expression by reverse transcriptase polymerase chain reaction (RT-PCR). After treatment with Gymnema sylvestre (GS) at indicated concentrations for 24 hours, the cells were harvested and the total RNA was extracted. The mRNA expression level of cytochrome c, caspase 3, PARP, ICAD, Bax, EGFR, and Bcl2 were detected by RT-PCR analysis (A). G3PDH worked as the equal loading control in this study. Fluorescence intensities of PCR bands were estimated by image J software (B).
The List of the Sequences of the Primers Used.

Abbreviations: PARP, poly(ADP-ribose) polymerase; ICAD, inhibitor of caspase-activated DNase; EGFR, epidermal growth factor receptor; G3PDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
In the present study, we observed that whereas GS had little or no cytotoxicity on normal liver cell line WRL-68, it had considerable apoptotic effect on A375 cells. One study 18 conducted earlier on rat also reported that GS had no or little cytotoxic effects on normal rat liver cells. The apoptosis triggered by GS in the present study was likely mediated through the activation of mitochondria-dependent cell death pathway; the latter was presumably triggered by the excessive ROS production, which in turn resulted in the loss of mitochondrial membrane potential. 19 On the other hand, a loss of proper balance between ROS production and generation of biochemical antioxidants could also lead to oxidative stress in all aerobic cells.20,21 An increase in the ROS production has long been implicated to the apoptotic response induced by several anticancer agents.22,23 The treatment with GS caused an increase in cellular ROS accumulation at an early hour of drug exposure (within 3 hours). Kang et al 24 also reported recently that GS has antioxidative property in diabetic rats. In the present study, ROS generation was found within 3 hours of drug exposure, after which we did not continue the study for any further period to find out its late effects. Therefore, loss of mitochondrial membrane potential in A375 cells at early hours might be responsible for triggering apoptosis. 25 Our results suggest that the accumulation of ROS in human melanoma A375 cells after treatment could possibly be responsible for the apoptotic response manifested by the cancer cells. However, loss of mitochondrial membrane potential is an important marker of apoptotic cells, 26 and our study also reveals the decrease in mitochondrial membrane potential from 47.0% (in control cells) to 7.9% (GS-treated cells) after 24 hours of incubation with 200 µg/mL of GS treatment. It is reported that cytochrome c released from mitochondria triggers caspase activation, which could be mediated by direct or indirect action of ROS. 27 The release of cytochrome c after GS treatment would further lend support to this contention as evidenced by the ELISA and RT-PCR studies. This result was also supported by the fact that pro-apoptotic mRNAs of caspase 3, Bax, and PARP genes were upregulated whereas the anti-apoptotic mRNAs of Bcl2 gene was downregulated. The altered ratio of the Bax/Bcl2 and the increase in caspase activity possibly led to the apoptosis of the A375 cells. Activated caspase 3 is known to cleave and upregulate some specific substrates, such as PARP proteins, 28 the activities of which were also detected in the GS-treated cells. To elucidate the possible mechanisms of apoptosis in A375 cells, we investigated the effects of GS with respect to mRNA expression by RT-PCR. PARP functions as a DNA damage sensor 29 and the increased expression of PARP supports the DNA breakage in apoptosis. Downregulation of ICAD indicates the active CAD molecule that can enter in the nucleus and degrade the chromosome. 30 Furthermore, we investigated the effects of GS on caspase 3 and PARP proteins expression by ELISA, in which similar trends of findings were observed to those of RT-PCR. GS treatment promoted cytochrome c, caspase 3, and PARP protein expressions dose dependently in the A375 cells. Decrease in mitochondrial membrane potential possibly induced altered expressions of caspase 3 and PARP proteins that could direct the cells toward apoptosis. 31
In this study, we found similar results of increased caspase 3 and PARP expression along with loss of mitochondrial membrane potential in drug-treated cells. Therefore, this is indicative enough that GS triggered apoptosis in A375 cells as a result of decreased mitochondrial membrane potential. Caspase 3 is known to actively take part in proteolytic cleavage of PARP protein. Increased expression of PARP was, therefore, also an indirect indication of greater extent of DNA degradation. Both these markers showed positive alteration in the drug-treated cells, hence caspase 3 and PARP have been considered here to represent significant markers of cells undergoing apoptosis. Taken together, these results suggest that the mechanism of GS induced apoptosis in A375 cells involves the activation of caspase 3 and PARP expressions. On the other hand, EGFR plays a novel role in cancer cell survival and proliferation. 32 We also found that GS reduced the expression level of EGFR that supported the antiproliferation activity of GS. Annexin V-FITC assay also indicates a higher percentage of apoptotic cells treated with GS. Percentage of apoptotic cells increased from 1.77% (in control cells) to 23.42%, 25.37%, and 31.89%, respectively, on 24 hours of incubation with GS. These results showed a clear indication toward the increased rate of apoptosis along with the higher dose of GS treatment. We also assessed the DNA damage by DNA fragmentation assay and fluorescence microscopic observations, which showed positive result toward ability of GS to induce DNA damage in A375 cells.
Conclusion
The antidiabetic property of GS had already been known, but its anticancer potentials had never been explored. This study clearly demonstrates that GS has considerable anticancer potential as well. Furthermore, the possible signal pathway of the drug has also been elucidated for the first time. The apoptotic pathway was linked to the mitochondria-dependent caspase route through caspase 3 and PARP activation, causing death to the cancer cells. The dual role played by this drug could be of interest in the design and formulation of specialized drugs.
Footnotes
Acknowledgements
The authors are grateful to Boiron Laboratories, Lyon, France for partial financial support of the work. Sincere thanks are due to Dr N. Boujedaini, Boiron Laboratories, for her encouragement and helpful comments.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported partly by a research grant sanctioned by Boiron Laboratories, Lyon, France to Prof A. R. Khuda-Bukhsh.