
Abstract
Intraperitoneal injection of amylin or its analog reduces Alzheimer’s disease (AD) pathology in the brains. However, self-injecting amylin analogs is difficult for patients due to cognitive deficits. This work aims to study the effects of amylin on the brain could be achieved by oral delivery as some study reported that amylin receptor may be present in the gastrointestinal tract. A 6-week course of oral amylin treatment reduced components of AD pathology, including the levels of amyloid-β, phosphorylated tau, and ionized calcium binding adaptor molecule 1. The treatment reduced active forms of cyclin-dependent kinase 5. Oral amylin treatment led to improvements in social deficit in AD mouse. Using immunofluorescence, we observed the amylin receptor complexed with the calcitonin receptor and receptor activity-modifying proteins in the enteric neurons. The study suggests the potential of the oral delivery of amylin analogs for the treatment of AD and other neurodegenerative diseases through enteric neurons.
Introduction
Amylin (also known as islet amyloid polypeptide or IAPP) is a highly conserved 37-amino acid peptide and is produced and co-secreted with insulin by β-cells in the pancreas. Amylin functions as a gut-brain axis hormone 1 -4 by binding to its receptor in the brain to reduce appetite and blood glucose. 5 -8 The amylin receptor (AmR) is a G-protein coupled receptor (GPCR) composed of calcitonin receptor (CTR) complexed with different receptor activity-modifying protein 1, 2 or 3 (RAMP1, 2 or 3). 9 , 10 Several studies have shown that AmR may exist in gastrointestinal (GI) tract, 11 , 12 however, which AmR subtype and in which cell type in the GI system are not clear. An amylin analog, pramlintide, was approved by the FDA and is a drug for type 1 and 2 diabetes. 13 Although pramlintide has a favorable safety profile in clinical use, 14 the drug has not been widely accepted by diabetes patients because of its delivery route (injection) given that these patients require separate injections of insulin. If AmR exists in GI track, pramlintide or other amylin drugs could be delivered orally to the patients. Amyloid-β peptide (Aβ) is the major component of AD pathology and share several features with amylin including binding receptors and degraded by the same enzymes. 15 Thus abundant Aβ may block the ability of amylin to bind its receptor in the AD brain. Amylin mediates brain functions including glucose metabolism and neural regeneration through the penetration of blood-brain barrier. However, high amount of amylin could aggregate in the type 2 diabetes. The aggregation may result in pancreatic cell mass loss and reduction of insulin secretion.
Several recent studies have demonstrated that peripheral treatment with human amylin or its clinically approved analog pramlintide 16 reduces the pathological cascade of Alzheimer’s disease (AD) and improves cognitive impairment in multiple mouse models of AD. 15,17,18 These studies strongly suggest that amylin analogs may become therapeutic agents for AD. 19 One concern is that, as dementia patients with AD have visuospatial impairments, they may be unable to administer the medication to themselves if only an injectable formulation of the drug is available. In addition, the extra burden for caregivers, who may be required to administer injections to the patients, also needs to be considered when developing amylin analogs for AD treatment.
In this study, we aimed to study whether and which subtype of the amylin receptor is expressed in the gastrointestinal tract. We hypothesized that if the amylin receptor is expressed in this region, administering amylin orally would reduce AD pathology in the brain. We found detected AmR expression in enteric neurons and demonstrated that like the intraperitoneal injection (i.p) of amylin, oral amylin delayed AD progression by reducing the accumulation of Aβ, the level of phosphorylated tau (pTau) protein and neuroinflammation in a mouse model of AD.
Materials and Methods
Mice and Experimental Treatments
AD model mice, heterozygous 5XFAD mice, which are amyloid precursor protein (APP)/presenilin 1 (PS1) double transgenic mice with 5 familial AD mutations, 20 were purchased from the MMRRC (Jackson Laboratory, Bar Harbor, ME, USA). All mice were maintained in a microisolator in the animal facility at Boston University School of Medicine, MA, USA. Human amylin was purchased from AnaSpec (Fremont, CA, USA). Female 5XFAD mice aged 3.5 months were used for the experiments (n = 10 in each treatment group). Amylin dissolved in water was freely administered to 5XFAD mice (200 μg/kg per day). Vehicle-treated mice drank only water and served as controls. The amylin dissolved in water and vehicle-treated water were changed every day. The mice had no change of body weight after 6 weeks post treatment compared to initial body weight, and there was no difference of body weight between the PBS treated and the oral amylin treated groups. Body condition will be monitored throughout the study according to the IACUC policy for post procedure monitoring. We will not allow mice to go Body Condition Scoring < 2 for minimizing suffering. All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Boston University Animal Care and Use Committee (PROTO201800192).
Immunofluorescence Analysis of the Small Intestines and Enteric Neurons of Mice
Small intestines were removed and fixed overnight in 4% PFA and then transferred to 30% sucrose in phosphate-buffered saline (PBS) at 4°C. The cryoprotected samples were embedded in OCT compound and sliced using a cryostat. Primary enteric neurons were fixed in 4% [wt/vol] PFA plus 4% [wt/vol] sucrose at 37°C for 15 min. The cryosections (∼15 μm) and neurons were blocked in 5% [wt/vol] BSA with 5% [vol/vol] donkey serum in PBS and incubated at 4°C overnight with Ramp1 (#1567 R), Ramp3 (#11972), CLR (#1860 R), and CTR (#0124 R) primary antibodies (1:400 dilution, Bioss, Woburn, MA, USA), an antibody against the neuronal marker TuJ1 (#ab18207, 1:1000 dilution, Abcam, Cambridge, MA, USA) and the nucleus marker DAPI. Fluorescent-labeled secondary antibodies (1:500 dilution, Alex Fluor Dye, Invitrogen, Carlsbad, CA, USA) were used in the immunoassays. Images were captured with a confocal laser-scanning microscope (Zeiss LSM 700) with a 63× oil objective lens and a 20× objective lens.
Primary Enteric Neuron Cultures
Explant cultures of the myenteric plexuses of newborn mice were established as described previously with minor modification (PMID 23975821). In brief, the intestines were removed by cutting the mesentery near the gut, and these pieces were stored in MEM-HEPES (pH 6.2; Invitrogen) on ice. The muscle layer was removed from the mucosa under a dissecting microscope. The muscle strips were collected and dissociated in a vial containing collagenase A (1 mg/ml, #11088785103, Roche Diagnostics, Indianapolis, IN, USA) in HBSS placed in an incubator (37°C, 5% CO2) for 20 min. After digestion, cells were obtained by centrifugation for 8 min at 356 × g at 4°C. The cells were placed in 0.01% trypsin solution at 37°C for 3 min, neutralized with FBS and dissociated into single cells. The cells were plated on glass coverslips precoated with Matrigel (1:200 dilution; Becton Dickinson Bioscience, Bedford, MA, USA) in complete enteric neuron medium (neurobasal A medium with B-27, 2 mM L-glutamine, 20 ng/ml epidermal growth factor, 20 ng/ml basic fibroblast growth factor, 1% penicillin/streptomycin/antimycotic, and 50 μg/ml gentamicin).
Purified neurons from isolated enteric cells were collected using the Neuron Isolation Kit (#130-115-389, Miltenyi Biotec, Bergisch Gladbach, Germany), and mouse non-enteric neurons were labeled with biotin-conjugated monoclonal antibodies and removed by retaining them within a column in a magnetic separator. The unlabeled enteric neuronal cells were collected and analyzed for further experiments.
Real-Time PCR
Total RNA was isolated from freshly dissected enteric neurons using RNA extraction kits (#74104, Qiagen, Hilden, Germany). cDNAs were obtained with SuperScript II reverse transcriptase using oligo-dT and random hexamer primers (#1708891, Bio-Rad, Hercules, CA, USA). An amplification reaction was prepared using SYBR Green reagent (#4309155, Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol. RT-qPCR was performed using a real-time fluorescence quantitative PCR instrument (ABI 7300, Applied Biosystems). The following specific PCR primer sets were used for RT-PCR analysis.

Immunohistochemical Analysis of Mouse Brains
Immunohistochemistry was used to characterize the pathology of mouse brains (n = 10 in each treatment group). Mouse brains were removed, post-fixed in 4% paraformaldehyde for 2 days, and treated with 30% sucrose in PBS for 2 days. Floating serial coronal cryosections (30 μm) were cut and stored at 4°C. 21 After washing with PBS, endogenous peroxidase activity was quenched by incubating the sections in 0.3% H2O2 in methanol for 30 min, and then the sections were washed in PBS.
The slides were preincubated in blocking solution (5% [vol/vol] goat serum [Sigma, St. Louis, MO, USA] and 0.3% [vol/vol] Triton-X 100 in PBS) for 1 hour at room temperature. The slides were incubated individually with primary antibodies overnight. For Aβ, mouse mAb 6E10 (1:1000, #803015, Biolegend, San Diego, CA, USA) was used, and for human pTau, AT8 (1:500, MN1020, Thermo Fisher, Waltham, MA, USA) was used. A mouse antibody against the microglial marker Iba-1 (1:500, #019-19741, Wako, Osaka, Japan) was used. The secondary antibodies used for immunohistochemistry were biotinylated mouse antibodies (1:4000, #PK-6101, #PK-6102, Vector Labs, Inc., Burlingame, CA, USA). Primary antibody binding was detected by biotin-conjugated secondary antibodies and a Vectastain ABC kit (#PK-4100, Vector Labs, Inc.) using DAB (3,3′-diaminobenzidine; Vector Labs, Inc.) as a substrate for peroxidase and counterstaining with hematoxylin. The end products were visualized as eight-bit RGB images using NIS Elements BR 3.2 software at total magnifications of 10× and 20×.
ImageJ software was used to analyze the immunostaining results. 18 ImageJ was used to measure the area of the plaques after adjusting for the threshold. The data from 3 independent investigators who were blinded to the treatment groups of all 3 sections were pooled and averaged.
Western Blotting
Western blot experiments of samples from each brain were replicated by 2 researchers in the laboratory to confirm the results. Total protein was extracted from the cortices and hippocampi of mouse brain with radioimmunoprecipitation assay (RIPA) buffer, and 1% Triton X-100 (TBS-x) buffer was used to extract the soluble fractions. 18 Fifty micrograms of protein were used to evaluate the protein levels of amyloid-β (Aβ), phosphorylated tau and p25-cyclin-dependent kinase (CDK) 5. To evaluate the expression of Aβ and amyloid precursor protein (APP), the blots were incubated with a mouse 6E10 antibody (1:1000; Biolegend) and an anti-APP-C99 antibody (1:2000, #MABN380, Millipore Sigma, Burlington, MA, USA), respectively. The levels of pTau and total tau in total protein samples were also determined using Western blotting with AT8 (1:1000, MN1020, Thermal Fisher) and total Tau (1:1000, sc-32274, Santa Cruz Biotechnology, Dallas, TX, USA) antibodies, respectively. The β-actin level was used as the control in the experiment.
We also examined the endogenous levels of tau proteins in total protein samples from 5XFAD mice. pTau levels were detected by Western blotting using CP-13 (monoclonal, 1:500 dilution) and PHF-1 antibodies (monoclonal, 1:500 dilution). The CP-13 and PHF-1 levels were quantified and compared. To evaluate the CDK5-p35-p25 pathway, Western blotting was used to detect CDK5 with a specific polyclonal antibody (1:2000, #2506 S, Cell Signaling Technology, MA, USA), and the proteins p35 and p25 were detected in brain extracts from 5XFAD mice with a specific monoclonal antibody against both proteins (1:2000 dilution, #2680 S, Cell Signaling Technology, Danvers, MA, USA). The levels of CDK5, p35 and p25 were quantified and compared.
Behavioral Tests and Data Analysis
The nest building test was performed as described in a previous study. 22 We acclimated all animals to the facility and home cages for least 2 weeks prior to the nesting experiment. For testing, individual mice were placed in microisolator cages (18 cm W × 19 cm L × 12 cm H) in a quiet testing room, 8 g of shredded paper nesting material (2 cm × 2 cm, Kitchen towel) was flattened on the bottom of the cage; and food and water were provided. The cages were left untouched for the 24-hour experimental period. The individual mice were carefully removed from the cages approximately 1 hour after the 24-hour period. We photographed each cage for later assessment. Nest construction was scored using the established system described by Deacon, which involves a 5-point scale. 22 In brief, the scores were as follows: (1) nestlet not noticeably touched (>90% intact); (2) nestlet partially torn (50%-90% intact); (3) nestlet mostly shredded but often no identifiable nest site; (4) an identifiable but flat nest; (5) a (near) perfect nest with a clear crater. During this assessment, photographs were scored by an investigator blinded to the genotypes of the mice.
Statistical Analyzes of the Mouse Experiments
The outcomes of the mouse experiments, including data on amyloid plaques, tauopathy and microglial cells, as well as the behavioral data, between the treatment and control groups were compared by using ANOVA followed (GraphPad software, San Diego, CA, USA) by Tukey’s test or Student’s t test. The mean ± SE and p value showing statistical significance are presented.
Results
Oral Amylin Treatment Reduces Amyloid Levels in the Brain
We first investigated whether oral amylin treatment can reduce the Aβ amyloid burden in the brain. We used 5XFAD mice aged 3.5 months, an age at which there are prominent Aβ amyloid deposits in the brain. The mice were given drinking water or human amylin (200 µg/kg) in 50 mL water daily for 6 weeks (Figure 1). Oral amylin treatment significantly reduced amyloid pathology in the cortex (p = 0.0105, t = 2.856, df = 18) and hippocampus (p = 0.0182, t = 2.597, df = 18) based on a measure of amyloid burden (average plaque size × plaque intensity) (Figure 1A). We next examined the effects of orally administered amylin on APP expression and processing in the brain. Oral amylin treatment reduced the levels of full-length APP protein (fAPP, p < 0.0001, t = 7.695, df = 18) but did not affect the levels of 1 form of secreted APP (sAPPα) in brain extracts (Figure 1B). Compared to control-treated mice, mice treated with oral amylin had lower levels of the APP processing fragments C99 (p = 0.0024, t = 3.526, df = 18) and Aβ oligomers (p = 0.0019, t = 3.643, df = 18). 23 Amylin treatment also reduced the levels of Aβ1-42 in the brain (p = 0.0412, t = 2.590, df = 6), as determined by ELISA; the effects of amylin on Aβ in the brain tended to be attenuated (Supplementary Figure 1).
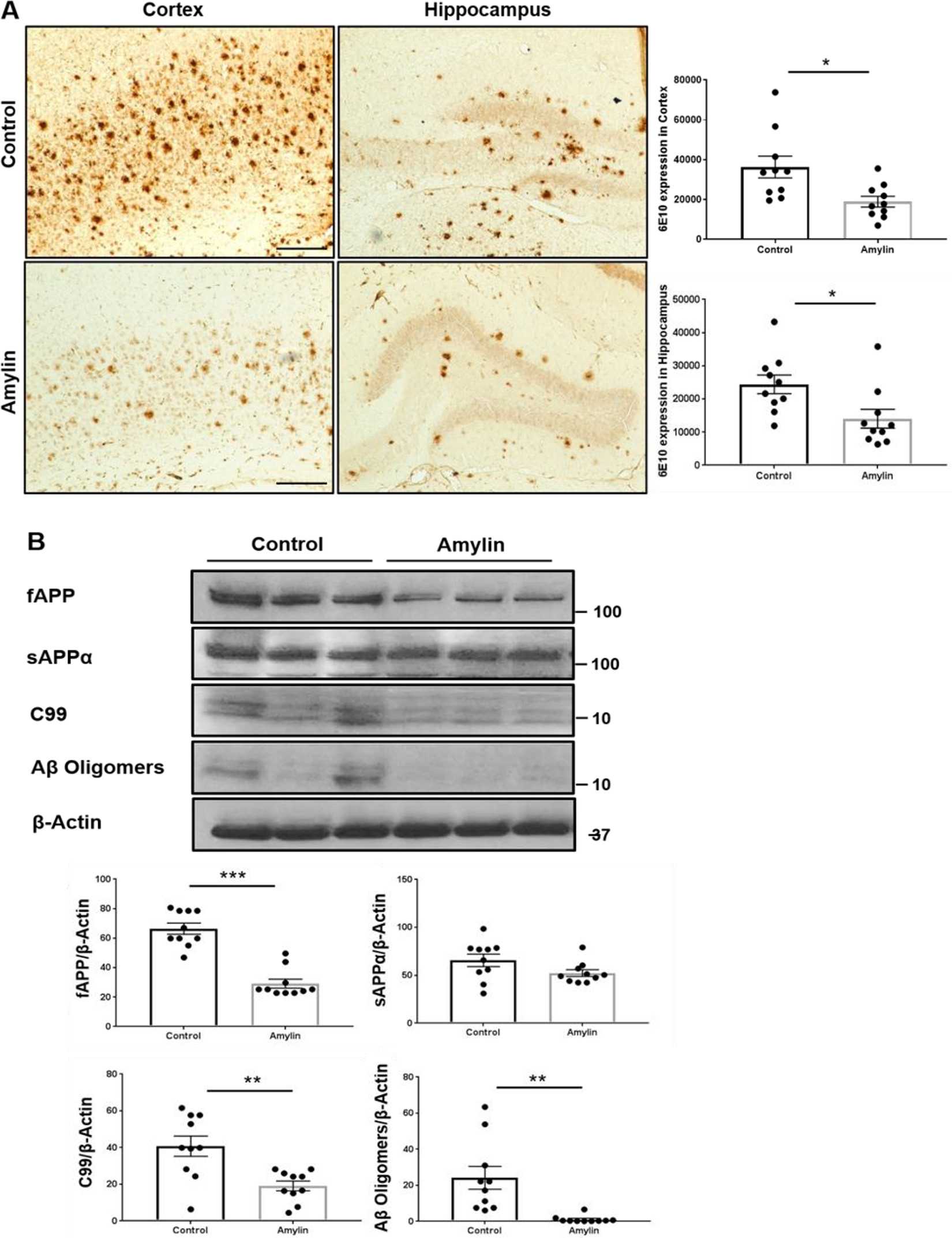
Oral amylin treatment reduces amyloid pathology in a mouse model of AD. Female 5XFAD mice aged 3.5 months (n = 10 for each treatment) were used and provided with amylin in water or water alone for 6 weeks. A, Brain sections were examined by using immunohistochemistry. Cortical and hippocampal sections were stained with an Aβ antibody (6E10) (scale bar: 200 μm). Compared to control treatment, amylin treatment significantly decreased the amyloid burden in the brains of 5XFAD mice (*p < 0.05). B, The expression of full-APP and secreted APPα, C99, and Aβ oligomers in brain tissues homogenate with TBS-x were detected by using Western blotting. All mice were examined, but a representative image was presented. Compared to water alone, oral amylin reduced the expression levels of all these proteins (**p < 0.01 and ***p < 0.001). For all comparisons, statistical analyzes (Student’s t test) were performed to compare the different groups.
Oral Amylin Treatment Reduces Tau Phosphorylation in the Brain
We next investigated whether oral amylin treatment can influence the other key component of AD pathology, phosphorylated tau (pTau), in the brains of 5XFAD mice. Immunostaining with an AT8 antibody (which detects the phosphorylation of both S202 and T205) revealed that oral amylin treatment strongly reduced tau pathology in the brain (p = 0.0304, t = 2.350, df = 18, Figure 2A). Next, western blotting of brain extracts from 5XFAD mice with 3 antibodies, namely, AT8, PHF1 (S396/404) and CP13 (S202), and normalization of the expression levels to those of actin showed similar effects of oral amylin treatment; the protein levels of phosphorylated tau protein were reduced (Figure 2B), but only blotting with the PHF1 antibody resulted in statistical significance (p = 0.0022, t = 3.577, df = 18). The changes in the levels of total tau did not reach statistical significance, which suggests that amylin selectively protects against tau pathology by decreasing the aggregation and phosphorylation of tau.

Oral amylin treatment decreases tau phosphorylation in a mouse model of AD. Female 5XFAD mice aged 3.5 months (n = 10 for each treatment) were used and provided with amylin in water or water alone for 6 weeks. A, Brain sections were examined by using immunohistochemistry. Cortical sections were stained with a phosphorylated tau antibody (AT8, S202 and T205) (scale bar: 200 μm). Compared to control treatment, oral amylin treatment significantly reduced pTau expression in the brains of 5XFAD mice (*p < 0.05). B and C, Total protein isolated from brain homogenates was evaluated for the expression of pTau (B) and p25-CDK5 signaling pathway-related proteins (C) by using Western blotting. Compared to control treatment, oral amylin treatment significantly reduced the protein expression levels of pTau, as detected by 3 different pTau antibodies, namely, AT8, PHF-1 and CP13 (**p < 0.01) and significantly reduced the levels of p35 and p25, but not CDK5 (*p < 0.05, **p < 0.01, ***p < 0.001). All mice were examined, but representative images were presented. For all comparisons, statistical analyzes (Student’s t test) were performed to compare the different groups.
The above data suggest that oral amylin selectively inhibits the signaling pathway(s) that phosphorylate tau. We examined the effects of oral amylin treatment on 2 pathways that are putatively involved in tau phosphorylation: 1) the p35-p25-CDK5 pathway and 2) the AKT-glycogen synthase kinase (GSK)3-β pathway. 24 Immunoblotting of lysates from the treated mice revealed that after normalization of the expression levels to those of actin, oral amylin treatment dramatically decreased the protein levels of p35 (p < 0.0001, t = 4.841, df = 18) and p25 (p = 0.0228, t = 2.489, df = 18), which are cleavage products associated with CDK5 activation (Figure 2C), but did not influence the expression level of CDK5. Like amylin injection, oral amylin treatment had little or no effect on the phosphorylation of AKT or GSK3-β (p = 0.143), suggesting that the actions of amylin were selective for the CDK5 pathway.
Oral Amylin Treatment Attenuates Neuroinflammation in the AD Brain
To determine the effects of oral amylin treatment on neuroinflammation in AD model mice, activated microglia were examined with immunostaining for ionized calcium-binding adaptor (Iba-1). 25 Oral amylin treatment significantly reduced the expression level of Iba-1 and the number of Iba-1+ microglial cells in the cortex of 5XFAD mice (p = 0.0256, t = 2.434, df = 18) and tended to do so in the hippocampi of 5XFAD mice (Figure 3A). In addition, we measured the levels of components of the Nuclear Factor kappa-light-chain-enhancer of activated B cells (NFκB) proinflammatory pathway and compared them (Figure 3B) between the oral amylin-treated 5XFAD mice and control 5XFAD mice. Compared with control treatment, oral amylin treatment significantly increased the protein level of IκBα (p = 0.0027, t = 3.479, df = 18) in brain extracts.
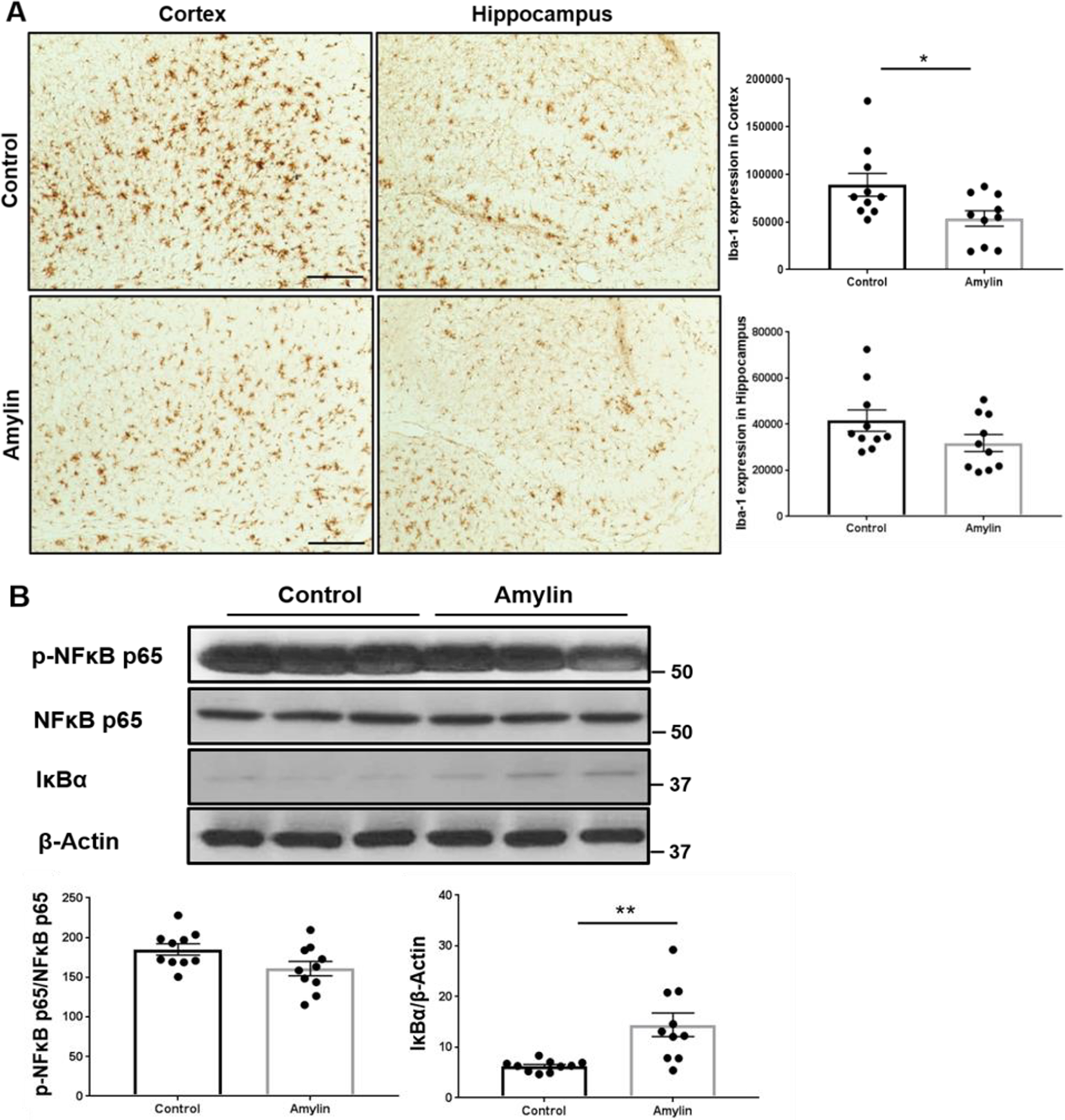
Oral amylin treatment reduces neuroinflammation in the AD mouse model. Female 5XFAD mice aged 3.5 months (n = 10 for each treatment) were used and provided with amylin in water or water alone for 6 weeks. A, Brain sections were examined for Iba-1 by using immunohistochemistry. Cortical and hippocampal sections were stained with an Iba-1 antibody (scale bar: 200 μm). Compared to control treatment, oral amylin treatment significantly reduced Iba-1 expression in the cortex (*p < 0.05). B, Brain homogenates from 5XFAD mice were evaluated for the expression of phosphorylated NFκB p65, total NFκB p65, and IκBα by using Western blotting. Compared to control treatment, oral amylin treatment significantly increased the level of IκBα (**p < 0.01). All mice were examined, but a representative image was presented. For all comparisons, statistical analyzes (Student’s t test) were performed to compare the different groups.
Oral Amylin Treatment Improves Learning and Memory in a Mouse Model of AD
Nest construction, which is a natural, species-typical behavior in rodents, was evaluated. Previous studies have reported that nest building, which is an indicator of well-being and social context in mice, decreases with age in rodent models of AD. 26 , 27 In this study, the nesting scores of in the 5XFAD transgenic mice in the nest building test were significantly lower than those of the wild-type mice (p < 0.001, F (1, 15) = 7.483). Oral amylin treatment significantly increased the nesting scores of the 5XFAD mice (Figure 4, p = 0.0059, F (1, 19) = 4.879), suggesting that oral amylin can improve both cognitive and social deficits in a mouse model of AD.

Oral amylin treatment improves cognitive and social function in a mouse model of AD. WT mice treated with drinking water and 5XFAD mice treated with drinking water alone or amylin in water (n = 6 in the WT group, n = 10 in the 5XFAD control group, n = 10 in the 5XFAD amylin-treated group) were subjected to a nest building test. 5XFAD mice had significantly lower nesting scores than WT mice (***p < 0.001). Compared to the control treatment, oral amylin treatment improved the cognition of 5XFAD mice, significantly increasing nest scores (### p < 0.001). For all comparisons, the mean ± SE is shown, and statistical analyzes (ANOVA followed by Tukey’s test) were performed to compare the different groups.
The Expression of the Amylin Receptor in Enteric Neurons
To confirm the existence of AmR and further characterize the receptor in GI track, we used immunohistochemistry to examine the expression pattern of AmR in the gastrointestinal (GI) tract. We found CTR and RAMP1 were strongly expressed in TuJ1-positive neurons in the small intestine but that RAMP3 expression was weaker in these neurons (Figure 5A). Since enteric neurons in the GI tract connect with the brain, especially the hypothalamus, the data suggest that AmRs, especially CTR/RAMP1, are mainly expressed in primary enteric neurons. To further confirm our observations, primary cells were isolated from longitudinal muscle/myenteric plexus preparations and were stained in vitro with the neuronal-specific marker TuJ1. As shown in Figure 5B, CTR and RAMP1 were mostly expressed in enteric neurons, whereas CLR and RAMP3 were widely distributed in other nonneuronal cells. Subsequently, the mRNAs were extracted from sorted neurons using microbeads with biotin-conjugated neuronal antibodies and analyzed for the quantification of different AmR-related genes (p = 0.0277, t = 3.384, df = 4). We found dramatically increased expression of the CTR gene in sorted enteric neurons (Ramp1 vs CTR, p = 0.0009; Ramp2 vs CTR, p = 0.0008; Ramp3 vs CTR, p = 0.0010; CLR vs CTR, p = 0.0008, F (4,15) = 10.83, p = 0.0002). These results indicated that AmR is expressed in the enteric nervous system and can bind amylin in the GI tract to communicate with neurons in the brain.

The amylin receptor (AmR) is expressed in enteric neurons. The small intestines and enteric neurons of wild-type (WT) mice were used. A, Small intestines were isolated from WT mice and double immunostained with antibodies against each component of the AmR (red) complex, either CTR, RAMP1, RAMP3 or calcitonin receptor-like receptor (CLR), and the neuron marker TuJ1 (green). DAPI was used to stain the nuclei. Line graphs of fluorescence profiles are shown for TuJ1-positive cells (scale bar: 10 μm). B, Enteric neurons were isolated and immunostained with antibodies against each component of the AmR complex, including CTR, RAMP1, RAMP3, and CLR, TuJ1 and DAPI. Fluorescence images of enteric neurons labeled with each antibody are shown (scale bar: 50 μm). C, The expression level of the Tubb3 gene (a neuronal marker) was significantly increased after the enteric neurons were enriched with neuronal-specific antibodies using microbeads (upper panel). Fold changes in the gene expression levels of AmR components, especially CTR, relative to those in unpurified samples and normalized to Gapdh are shown. At least 3 experiments were performed. *p < 0.05, ***p < 0.001; Student’s t test; one-way ANOVA followed by Tukey’s multiple comparisons test. The data are represented as the mean ± SEM.
Discussion
Although pramlintide and other amylin agonists may be potential therapies for AD, pramlintide is delivered by subcutaneous injection for diabetes patients; however, self-injection of the drug is difficult or impossible for AD patients, and thus the administration of pramlintide increases the burden on caregivers. This study demonstrated that amylin can be delivered orally to reduce the AD-related pathological cascade in the brain, probably through AmR in enteric neurons in the GI tract.
Figure 5 shows that enteric neurons, but not other types of cells in the GI tract, mainly expressed AmR, complexed with CTR and RAMP1. Another study showed that CTR is expressed in enteric neurons during maturation. 28 Since enteric neurons connect the gastric intestinal system with neurons in the brain, especially the hypothalamus, 29 the data suggest that the therapeutic effects of oral amylin on AD were probably mediated by AmR on enteric neurons. Heterodimers of CTR and either RAMP1 or RAMP3 preferentially bind amylin as well as the amyloid-β (Aβ) peptide, and the brain predominantly expresses both RAMP1 and RAMP3. 16,30,31 It has been shown that microglial cells express only RAMP3, while other neuronal cells express both RAMP1 and RAMP3. 32 RAMP1 has an impact on the immune system. For example, RAMP1 knockout results in a dysregulated immune response; in contrast, RAMP3 knockout mice appear normal until old age, suggesting that RAMP3 knockout is relatively innocuous. 33
It is unclear how amylin binds enteric neurons affect microglia in the brain. However, several studies have shown that central nervous system (CNS) and enteric nervous system (ENS) share mechanisms of neuroimmune interaction. For example, CX3CR1 is highly expressed in ENS, but the receptor is implicated in neuron-microglia interaction. 34 In addition, condition change of intestine may result in the modification of neuron and glia in brain regarding neuroimmune reactions. 35 Therefore oral administration of amylin affects their receptors in ENS, and the amylin function in CNS could be increased and decreased inflammation through neuron and immune cells.
The current study (Figures 1 –4) demonstrates that oral amylin treatment reduces multiple types of pathology found in the AD brain, including amyloid pathology, aggregated p-tau and neuroinflammation, suggesting that the benefits of amylin-type peptides extend beyond a simple amyloid-centric mechanism. This is similar to the benefits of intraperitoneal amylin administration in AD models, which are mediated by AmR and result in improvements in learning and memory. 15 Amylin is fully degraded by proteases secreted by pancreas into small intestine. Although extremely low pH environment, like in stomach, can cause hydrolysis of proteins non-enzymatically, it is extremely slow. Thus, most orally taken amylin peptides probably pass into small intestine to bind to AmR 36 or are degraded if not bind to AmR. Our studies of the 2 main pathological pathways linked to tau, the CDK5 and GSK3β pathways, showed selective inhibition of the p35-p25-CDK5 pathway (Figure 2). 37 -39 We did observed that oral amylin treatment reduced AT8 immunostained with pTau in the brain (Figure 2A), however, western blot only showed tendency but did not reach statistical significance (Figure 2B). Previous experiments confirmed that AT8 antibody showed the difference of pTau expression in sarkosyl-insoluble fraction by amylin i.p injection. However, in this study we used oral amylin, which was less effectively compared to i.p amylin, and we used total protein extraction. Nevertheless, we observed that another pTau antibody, PHF1, showed the reduction of pTau in total protein fraction by oral amylin (Figure 2B). In addition, NF-kB could not explain proinflammatory pathways in whole brain. However, we clearly found the induction of IkB (Figure 3B), especially IkBα has a major role regarding the NF-kB with other IkB isoforms to block the NF-kB activation. We also found that Iba-1 expression in the brain was reduced by oral amylin treatment. The pathophysiology of AD is clearly pleiotropic, yet most attempts at disease modification targeting a single component of AD pathology have failed in clinical trials. 40 -42 Taken together, our results suggest that oral amylin antagonized degenerative pathways linked to AD, providing neuroprotection of learning and memory, probably through the binding of amylin receptor on enteric neurons in the GI tract.
Supplemental Material
Supplemental Material, sj-docx-1-aja-10.1177_15333175211012867 - Oral Amylin Treatment Reduces the Pathological Cascade of Alzheimer’s Disease in a Mouse Model
Supplemental Material, sj-docx-1-aja-10.1177_15333175211012867 for Oral Amylin Treatment Reduces the Pathological Cascade of Alzheimer’s Disease in a Mouse Model by Hana Na, Hua Tian, Zhengrong Zhang, Qiang Li, Jack B. Yang, Liam Mcparland, Qini Gan and Wei Qiao Qiu in American Journal of Alzheimer's Disease & Other Dementias
Footnotes
Abbreviations
AD, Alzheimer's disease; AmR, amylin receptor; APP, amyloid precursor protein; Aβ, amyloid-β; CTR, calcitonin receptor; CLR, calcitonin receptor-like receptor; CDK, cyclin-dependent kinase; GI, gastrointestinal; GSK, glycogen synthase kinase; GPCR, G-protein coupled receptor; I?B, inhibitor of ?B; Iba, ionized calcium-binding adaptor; NF?B, nuclear factor kappa-light-chain-enhancer of activated B cells; PBS, phosphate-buffered saline; pTau, phosphorylated tau; RAMPs, receptor activity-modifying proteins; TBS, triton X-100 in PBS; WT; wild-type.
Authors’ Note
Hana Na and Hua Tian contribute equally to this manuscript.
Acknowledgments
HN designed the experiments, analyzed data, and co-wrote the manuscript, HT designed the GI tract and behavior experiments and analyzed data, ZZ performed the primary enteric neuron experiment, QL performed the mice experiments, analyzed data, JBY performed immunostaining and western blot assays and analyzed data, LM performed immunostaining and western blot assays and analyzed data, QG performed the behavior experiments, injection, WQQ designed and supervised the study and analysis and co-wrote and edited the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Alzheimer’s Disease Association (IIRG-13-284238); NIA (R21AG045757, RO1AG-022476, K24AG050842, R56AG059805 and RO1AG059424); and the Ignition Award (W.Q.Q.). Hua Tian was supported by National Natural Science Foundation of China (81803757).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.