
Abstract
By incorporating appropriate drug(s) into lipid (biobased) nanocarriers, one obtains a combination therapeutic for dementia treatment that targets certain cell-surface scavenger receptors (mainly class B type I, or “SR-BI”) and thereby crosses the blood-brain barrier. The cardiovascular risk factors for dementia trigger widespread inflammation -- which lead to neurodegeneration, gradual cognitive/memory decline, and eventually (late-onset) dementia. Accordingly, one useful strategy to delay dementia could be based upon nanotargeting drug(s), using lipid nanocarriers, toward a major receptor class responsible for inflammation-associated (cytokine-mediated) cell signaling events. At the same time, the immune response and excessive inflammation, commonly observed in the very recent human coronavirus (COVID-19) pandemic, may accelerate the progression of brain inflammatory neurodegeneration—which increases the probability of post-infection memory impairment and accelerating progression of Alzheimer’s disease. Hence, the proposed multitasking combination therapeutic, using a (biobased) lipid nanocarrier, may also display greater effectiveness at different stages of dementia.
Background
As stated in several reviews over the past 2 decades, there has been interest in using nanoparticles as an approach for the formulation of poorly (water-)soluble drugs [e.g., 1 -3 ]. In addition, nanoparticles can be formulated to provide passive or even active targeting, for drug delivery, when injected intravenously [e.g., 2 ]. As one example, surfactant-coated nanoparticles have been reported by Petri et al 4 to considerably enhance the antitumor effect of doxorubicin against intracranial glioblastoma in rats. [Their analysis, of plasma protein adsorption on the surface of the drug-loaded nanoparticles by 2-dimensional electrophoresis, revealed that the surfactant coating on the nanoparticles induced a considerable adsorption of apolipoprotein A-I (apoA-I).] These authors contend that the considerably enhanced antitumor effect may result from the interaction of apoA-I, which itself is anchored (i.e., adsorbed) onto the surfactant-coated nanoparticles, that is favored toward the “scavenger receptor class B type I” (SR-BI) located at the blood-brain barrier (BBB). 4
The foregoing findings fit well with the situation described elsewhere (e.g., 1 ) regarding certain “actively targeted” lipid nanoemulsions (namely, the Filmix® nanoemulsion as well as a small group of other (protein-free) parenteral lipid nanoemulsions); the SR-BI receptor emerged as the most likely candidate receptor for primary involvement in ligand-receptor binding of some of these (surface-active lipid) nanoemulsions at selected target cells, including many tumors. More specifically, the same foregoing findings are consistent with published conclusions (cf. 1 ) that SR-BI emerges as the main candidate facilitating enhanced endocytosis of “lipid-coated microbubble/nanoparticle-derived” (LCM/ND) nanoemulsions (containing mostly dispersed lipid cubic-phase nanoparticles—see Figure 1) into various target cells. Moreover, in this particular nanoparticle drug-delivery approach, the self-assembled lipid “nanoparticle structure” itself (upon injection into the bloodstream) is apparently successfully utilized as the “active” targeting ligand (see Filmix® nanoemulsion below) directed toward the appropriate receptors on the target-cell surface (cf. 1 ).
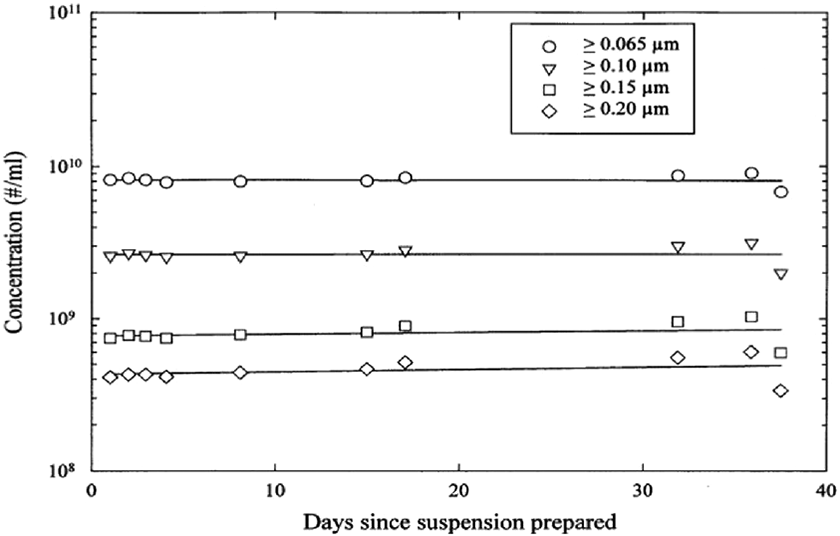
LCM/ND nanoemulsion stability over time. (Adapted from ref. 89).
Lipid Cubic Phases as “Actively Targeted” Nanoemulsion Drug-Delivery Vehicles
In a 2008 study by Salentinig et al, 5 a (shearing) procedure is described for the preparation of stable dispersions of nanostructured lipid mesophases (comprising “surfactant lipids,” which were mostly monoglycerides). These authors report that it was possible to produce high quantities of internally self-assembled nanoemulsion particles of controlled size. The dispersed particles containing different well-ordered nanostructures are kinetically stabilized systems, which their study shows are stable for several months. 5 They further explain in this study that such nanoemulsion particles, containing inverse-type liquid-crystalline phases, continue to attract much attention currently due to their great potential for applications in pharmaceutical, cosmetic, and food industries. [For example, the authors point out that the formulation of such nanostructured systems is attractive due to the possibility of combining the solubilization capacity for lipophilic drugs with their controlled release 5 (cf. 6 - 12 ).] In particular, an advantage of these nonlamellar liquid-crystalline nanostructures, such as inverse cubic phases, is the increased surface area generated by their inherent nanostructure 13 (cf. 14 ). Such nanostructured lipid mesophases are also much more robust when compared to liposomal delivery technologies by virtue of their semirigid periodicity. 13 Moreover, an important attribute of a limited number of lipid-based liquid-crystalline systems is that they are colloidally stable in excess water (e.g., 13, 15 ), thereby allowing for the predispersion of such liquid-crystalline systems in blood plasma in the form of submicron particles suitable for intravenous drug delivery. Polar lipids which are known to exhibit such phase behavior include monoglycerides, 15 cholesterol, and cholesterol esters 16 —all of which are repeatedly described as occurring in substantial concentrations within, the “particularly preferred form” of, LCM/ND nanoemulsion formulations. 17 - 25
As summarized in several reviews of nanotechnology-based drug-delivery systems, active targeting often involves conjugating the nanoparticle to an actual targeting moiety, thereby increasing the delivery of a drug to a target via the use of specific interactions at target sites. Such cell-surface interactions can represent antibody-antigen, lectin-carbohydrate, or ligand-receptor binding. 26 - 28 Yet, in this nanoparticle drug-delivery approach, sometimes the self-assembled “nanoparticle structure” itself can be utilized as the effectively “active” targeting ligand directed toward certain receptors on the target cell surface. [Note that the documented similarities in lipid composition, between naturally occurring high-density lipoproteins (HDL) versus the artificial biomimetic (LCM/ND-nanoemulsion) nanocarrier particles, can partially simulate or mimic the known heterogeneity (i.e., subpopulations or subspecies) of HDL particles. 1 Such biomedical application of colloidal drug-nanocarriers can potentially be extended to the treatment of complex medical disorders like dementia.] The above description or picture regarding active targeting, via ligand-receptor binding, fits well with the conclusions expressed earlier 1,27 concerning lipoprotein receptor-mediated drug delivery to tumor cells via targeted LCM/ND nanoemulsions. Specifically, the surface-membrane receptor known as “scavenger receptor class B type I” (or SR-BI) emerged as the most likely candidate target (of all lipoprotein receptors), with its documented multiligand capability (cf. 1 ), for primary or major involvement in enhanced endocytosis of LCM/ND nanoemulsions into various target cells. Upon binding of such lipid nanoemulsion particles to the target cell-surface receptors, dissociation of the drug from the nanoemulsion particle can occur either at the (adjacent) extracellular space, or at the cell surface, or intracellularly. 1 Also, the multiligand capability of SR-BI (in rodent), also known as CLA-1 (in human), is well documented elsewhere (cf. 1,29 - 33 ).
Biomimicking of Targeting Behaviors of “Reconstituted Lipoprotein” Vehicle(s)
As reviewed by Lacko et al, lipoproteins have long been considered to have structural properties that enable them to serve as drug delivery vehicles—because of their ability to incorporate hydrophobic drugs into their hydrophobic cores, and subsequently facilitate the cellular uptake of drugs via receptor-mediated mechanisms. 34,35 These authors, however, further point out that the utilization of native HDL as a drug-delivery vehicle is fraught with difficulties, namely the difficulty of large-scale isolation, biosafety issues and, consequently, the excessive cost involved in the development of the HDL-drug formulations. 35 This impracticality was unfortunate since the SR-BI receptor is known to be a major receptor for HDL (with their major apolipoprotein (apo)A-I). Moreover, Fung et al 36 specifically found that SR-BI mediates the uptake and transcytosis of HDL across brain microvascular endothelial cells (i.e., across the BBB). These investigators further argue that manipulation of HDL transcytosis across the BBB to increase delivery of plasma apoA-I may, in turn, facilitate increasing the transport of “HDL-like synthetic particles” containing therapeutic drug across the BBB to treat neurodegenerative disorders such as Alzheimer’s disease. 36
In view of the above difficulties associated with native lipoproteins, alternative strategies have been developed via artificially prepared “reconstituted lipoprotein(s)” for the purpose of targeted drug delivery. The continuing motivation to pursue such reconstituted lipoproteins stems from the expectation that lipoproteins increase the therapeutic efficacy by targeting drug(s) to tumors and other target tissues that overexpress lipoprotein receptors and, thus, minimize systemic toxicity (by shielding the drug from contact with most normal tissues 35 ). Upon entering the blood circulation, the delivery of the drug from the reconstituted lipoprotein nanoparticle is facilitated by a specific receptor/ligand interaction, provided by the particular polypeptide component (apolipoprotein, e.g. apoA-I) of the reconstituted-lipoprotein complex. 31,34,37,38 As concerns the lipoprotein receptor itself, a likely candidate is CLA-1/SR-BI which is often found to be overexpressed on many brain-tumor cells and neurodegenerative tissues in comparison to normal tissues (cf. 1,34,35 ). Note that the “reconsituted lipoprotein vehicle” was at first constructed solely of lipids, whereas, aoplipoprotein(s) (needed for targeting) were acquired only after incubation with serum. This concept of a pure-lipid nanocarrier, which can successfully acquire apolipoprotein(s) upon contact with blood plasma, is similarly described elsewhere in the literature. For example, Williams and Scanu 39 report that phosphoglyceride liposomes, injected intravenously, pick up endogenous apolipoprotein A-I (aopA-I); morover in vitro, it has been found that phosphoglyceride liposomes incubated with plasma acquire apoA-I at the expense of HDL 39 (cf. 40 ).
An analogous situation is believed to exist in the case of LCM/ND nanoemulsions. Specifically, the lipid composition of the LCM/ND nanoemulsion type (consisting of various glycerides, cholesterol, and cholesterol esters) is similar to lipids contained in several plasma lipoproteins (i.e., resembles the lipid content of a “generic” lipoprotein 31,35 ). Accordingly, when LCM/ND nanoemulsion(s) are injected into the bloodstream, it is believed that they likely acquire (i.e., bind) plasma apolipoprotein(s). Consistent with all of the above considerations, it was previously proposed that such bound species of apolipoproteins (e.g., apoA-I) are recognized by the corresponding lipoprotein receptors (often found overexpressed on the surface membrane of various target cells) ( 1 ; also see below).
Calcium Dyshomeostasis, and the Amyloid-β Ion Channel Hypothesis of Alzheimer’s Disease
Past mammalian and human studies (e.g., 41 ) have shown that low-grade inflammation and endothelial dysfunction contribute to reduced information processing speed and executive functioning in an older population. These interacting processes involve pathophysiological cascades which lead to neuronal (intracellular) Ca2+ increase, neurodegeneration, gradual cognitive/memory decline, and eventually dementia.
As explained in many reviews 42 - 45 by different investigators, it has been recognized for over 2 decades that disturbance of the intracellular calcium homeostasis is central to the pathophysiology of several neurodegenerative disorders. As concerns Alzheimer’s disease, it is believed by many researchers that enhanced calcium load may be brought about by extracellular accumulation of amyloid-β (Aβ) in the brain. Such studies have laid the foundation for the popular idea that amyloid-β peptides (39-42 amino acid molecules) are, in part, toxic to brain tissue because they form aberrant ion channels in cellular membranes and thereby disrupt Ca2+ homeostasis in brain tissue and increase intracellular Ca2+. More specifically, later studies indicated that soluble forms of Aβ facilitate influx through calcium-conducting ion channels in the plasma membrane, leading to excitotoxic neurodegeneration. 42,43
The precise cellular pathway(s) by which the amyloid-β peptides bring about excitotoxic neurodegeneration has been much debated. A common cellular picture used to explain the disruptive effect of calcium dyshomeostasis within brain tissue, appearing often in the literature (e.g., 46,47 ), involves a central role for the tripartite glutamatergic synapse in the pathophysiology of Alzheimer’s disease. Glutamate is the primary excitatory neurotransmitter in the brain and plays an important role in cognition and memory, but alterations in glutamatergic signaling can lead to excitotoxicity. This “Ca2+ dyshomeostasis”-induced excitotoxicity occurs when uncontrolled glutamate release surpasses the capacity of astrocytic clearance mechanisms, and is linked to several neurodegenerative disorders including Alzheimer’s disease 46 (cf. 47 ). (More generally, it should also be noted that various other alterations of intracellular signaling can lead to neurovascular degeneration. For example, Calabrese and coworkers 48 describe the major pathogenic factors involved in vascular cognitive impairment, emphasizing the relevance of cerebrocellular stress and neurohormetic responses to neurovascular insult. Similarly, this research group has recently 49 discussed various cellular mechanisms (e.g., oxidant/antioxidant status, oxidative stress, and the vitagene network) underlying Alzheimer’s-disease neuroinflammatory pathogenesis that are contributory to Alzheimer’s disease. 48 - 50 )
Historical support for the above amyloid-β ion channel hypothesis, or so-called “calcium hypothesis,” has also been observed at the clinical level. 51 Namely, there is little correlation between the amounts of fibrillar (insoluble) deposit at autopsy and the clinical severity of Alzheimer’s disease. In contrast, a good correlation exists between early cognitive impairment and levels of soluble forms of Aβ in the brain. 52 (Aggregation of Aβ proceeds from formation of soluble (low molecular weight) spherical oligomers toward eventually assuming a final and stable conformation as insoluble fibrils from which amyloid-β plaques are constituted.) Hence, neurotoxicity is associated with soluble aggregates (i.e., oligomers) of Aβ rather than with the plaques themselves. Accordingly, related experimental work has already shown that application of soluble Aβ oligomers (but not monomers or fibrils) to cultured neuroblastoma cells evoked large increases in cytosolic calcium that arise largely through Ca2+ influx across the plasma membrane. 52
As summarized by Di Scala et al, 51 the structure of amyloid pores has been extensively studied by ultrastructural methods. In particular, one group of investigators recently applied strategies (widely used to examine the structure of membrane proteins) to study the 2 major Aβ variants, namely, Aβ (1-40) and Aβ (1-42). Under the optimized detergent-micelle conditions: 1) Aβ (1-40) aggregated into amyloid fibrils; 2) contrariwise, Aβ (1-42) assembled into oligomers that inserted into lipid bilayers as well-defined pores. 53 (These amyloid pores adopted characteristics of a β-barrel arrangement.) Because Aβ (1-42), relative to Aβ (1-40), has a more prominent role in Alzheimer’s disease, the higher propensity of Aβ (1-42) to form β-barrel pore-forming oligomers is an indication of their importance in Alzheimer’s disease. 53 Recently, a different research group reported very similar findings. 54 As background for their study, these latter authors point out that: i) elevated Aβ (1-42) plasma levels have been correlated with the progression of late-onset forms of Alzheimer’s disease; ii) Aβ (1-42) is significantly more neurotoxic than Aβ (1-40) both in vivo and in neuronal cell culture; and iii) memory impairment is believed to be driven by Aβ (1-42) disruption of long-term (hippocampal) potentiation. In accordance with these considerations, the authors’ own detailed experimental data 54 indicated that Aβ (1-42) assemblies in oligomeric preparations form ion channels (in membranes excised from cells of neuronal origin). In contrast, Aβ (1-40) oligomers, fibrils, and monomers did not form channels. Moreover, ion channel conductance results suggested that Aβ (1-42) oligomers, but not monomers and fibrils, formed pore structures. The authors concluded that their findings demonstrate that only Aβ (1-42) contains unique structural features that facilitate membrane insertion and channel formation, now aligning ion channel formation with the neurotoxic effect of Aβ (1-42) compared to Aβ (1-40) in Alzheimer’s disease. 54
This neurotoxic effect of Aβ(1-42) in Alzheimer’s disease is further supported by neuropathology findings. As reviewed recently by Liu et al, 55 immunogold electron microscopy has been carried out which demonstrates that Aβ(1-42) can be found in multivesicular bodies of neurons in the human brain, where it is associated with synaptic pathology. In addition, much evidence suggests that Aβ oligomers are more potent than Aβ fibrils in eliciting abnormalities in synaptic functions. Oligomers of Aβ(1-42) are considered the most synaptotoxic forms, responsible for early cognitive deficits in Alzheimer’s disease. 55,56 Accordingly (and also consistent with the mention of “neuronal (intracellular) Ca2+ increase,” “excitotoxicity,” and “neuroinflammatory pathogenesis” at the start of this Section [paragraphs 1-3]), the resultant pathological level of Ca2+ signaling leads to gradual loss of synaptic function and ultimate neuronal cell death, which correlates clinically with the progressive decline in cognition/memory and the development of pathological neural tissue seen in Alzheimer’s-disease patients. 55
Inflammasomes, Gut-Brain Axis, Serum Amyloid A (SAA) Versus SR-BI Targeting, and Cognitive Impairment
Besides the above considerations about amyloid pore formation and synaptic pathology, systemic inflammation is well known to increase the risk for cognitive decline in human neurodegenerative disorders including Alzheimer’s disease. This belief is based on wide acceptance that several factors -- including obesity, diabetes, acute injuries, aging, and neurodegenerative disease -- can trigger a sustained immune response in the central nervous system (CNS) leading to neuronal dysfunction and destruction by microglia activation and release of neuroinflammatory mediators (e.g., 1,41,57 ). Initiation of the inflammatory response by brain microglia involves the intracellular multiprotein complexes termed “inflammasomes” (e.g., 57 - 59 ). The “NLRP3 inflammasome” has become the most studied inflammasome, over recent years, due to its activation by a diverse range of stimuli and contribution to the pathology of inflammatory diseases. 60 - 64 More specifically, NLRP3 inflammasome activation has been implicated in the pathogenesis of neurodegenerative diseases, especially Alzheimer’s disease; various findings suggest that NLRP3 inflammasome activation, induced by amyloid-beta (Aβ) peptide material, promotes the pathogenesis of Alzheimer’s disease by triggering the release of proinflammatory cytokines and neuroinflammation. 60,62 However, animal model studies demonstrate that HDL protects against memory deficits, neuroinflammation, and cerebral amyloid angiopathy; recently, in vitro studies confirm that HDL reduces vascular Aβ accumulation and attenuates Aβ-induced endothelial inflammation. 65 Correspondingly, in cellular experiments where the NLRP3 inflammasome was inhibited, it has been reported that an upregulation of SR-BI receptor expression was observed (but there was no effect on the expression of scavenger receptor class A). 66 Since SR-BI is known to act as a receptor for both HDL/apoA-I and SAA [see below], it is consistent that SAA has been detected in Alzheimer’s-disease brain and this finding supports a linking of SAA with CNS pathophysiology. 67 Lastly, cerebrovascular disease is a common co-morbidity in patients with Alzheimer’s disease, and is believed to exacerbate the cognitive impairment as well as to lower the threshold for developing dementia. Moreover, a dysregulated hemostasis and chronic vascular inflammation further impede vascular function, where its associated mediators interact synergistically. In Alzheimer’s disease, the cerebral vasculature is the location where multiple pathogenic processes converge and contribute to cognitive impairment. When inflammatory cytokines and chemokines reach the brain parenchyma, they can be neurotoxic, and spur local inflammation by activation of glia. In addition, this activation of glia cells can increase Aβ secretion and cleavage. Exposure of endothelial cells to Aβ will itself, in turn, promote the release of a large variety of inflammatory mediators. 68
Particularly noteworthy is more recent research 69,70 indicating that chronic inflammatory stimulus in the gut may induce (e.g., via serum amyloid A (SAA)) the release of proinflammatory cytokines. At the same time, increased BBB permeability due to aging (or dysfunction), in turn, allows these proinflammatory cytokines to enter the brain, inducing glia reactivity. 69,70 These recent findings and various past studies indicate that inflammation plays an important role in the process of Aβ deposition and, therefore, the inhibition of inflammatory cascades may attenuate amyloidogenic processes—such as Alzheimer’s disease 71 (cf. 72 ). Hence, an effective preventive and therapeutic strategy could be based upon nanotargeting drug(s) toward a major SAA receptor responsible for numerous SAA-mediated cell signaling events leading to cognitive decline and eventually Alzheimer’s disease or (late-onset) dementia.
Specifically, earlier research 30 has already confirmed that SR-BI receptors (or its human ortholog CLA-1) function as cell-surface SAA receptors—which bind, internalize, and mediate SAA-induced proinflammatory effects (cf. 73 ). However, Baranova et al additionally report that (in cell culture) ligands for CLA-1/SR-BI “efficiently compete” with SAA for CLA-1/SR-BI binding. 30 (For example, it has already been documented in the literature that both apoA-I and SAA are substrates for SR-BI, which indicates that SR-BI could mediate the transport of both proteins across the BBB (e.g., 74 )). Not surprisingly, therefore, Robert et al have recently asserted that many lines of evidence suggest a protective role for HDL and its major apolipoprotein (apo)A-I in Alzheimer’s disease. 75 Accordingly, a similar benefit (of “competitive binding” to SR-BI receptors) may well accompany the clinical intravenous use of the (“HDL-like”) LCM/ND lipid nanoemulsion vehicle—which has already been repeatedly described in the peer-reviewed literature (based upon numerous in vivo animal studies) as a targeted, apoA-I-based, (SR-BI mediated) drug-delivery agent. 1 Moreover, by incorporating drug molecules into the LCM/ND lipid nanoemulsion type, one is likely to obtain a multitasking “combination therapeutic” capable of targeting cell-surface SR-BI. This (intravenous) colloidal-nanocarrier therapeutic would make it possible for various cell types, all potentially implicated in Alzheimer’s disease 76,77 and/or (late-onset) dementia, to be simultaneously sought out and better reached for localized drug treatment of brain tissue in vivo. 1,45,78,79
Past Targeted Nanotherapy Using “HDL-like” (Biomimetic) Lipid Nanoemulsion
The earlier-described LCM/ND lipid nanoemulsion type is well documented 1,45,76 - 79 to be useful for the highly selective delivery of (easily incorporated) lipophilic dyes, labels, or low-molecular-weight drugs to various types of solid tumors and certain other (noncancerous) hyperproliferative-disease lesions/sites. All these lesions consistently display an increased (cell-surface) expression and/or activity of lipoprotein receptors, including, notably, the (class B) scavenger receptor known as SR-BI (or sometimes as CLA-1, the human SR-BI ortholog). Such data on SR-BI expression and function are noteworthy; namely, SR-BI has emerged as the lipoprotein receptor primarily involved in the enhanced endocytosis (i.e., enhanced intracellular uptake) of LCM/ND lipid nanoemulsions into hyperproliferative-disease sites. 1,45,76 - 79
First, as concerns tumors, an independent evaluation of this type of lipid nanoemulsion has appeared in a review article by Constantinides et al. 80 At the same time, this particular study provides certain relevant data that are useful as a test of the expectation that a significantly enhanced endocytosis of LCM/ND lipid nanoemulsion (likely mediated by SR-BI) ought to be readily detectable in Hep3B human hepatoma cells. This expectation arises from the fact that SR-BI expression, which is well described for HepG2 cells, has also been documented in Hep3B cells. Furthermore, when studying the effect of chemical agents causing decreased SR-BI levels in Hep3B hepatoma cells, the same chemical agents were observed to cause decreased uptake of HDL lipids into Hep3B cells (for a review see ref. 1 ). In actuality, a noticeably enhanced uptake of this (dye-carrying) LCM/ND lipid nanoemulsion type into varied tumor cells is reported by Constantinides et al 80 and, as expected, the observed enhanced uptake is particularly marked in Hep3B hepatoma cells (see Table 24.1 in ref. 1 ; cf. “parent Table” (p. 763) in Adv. Drug Deliv. Rev. (2008) 60:757–767). The LCM/ND lipid nanoemulsion version employed by these authors is called Emulsiphan. Most solid tumors displayed enhanced uptake of this Emulsiphan version of (dye-labeled) LCM/ND lipid nanoemulsion; however, these tumors did not do so to the same degree. Nonetheless, it is noteworthy that all of the varied tumor cells listed in Table 24.1 (of 1 ; cf. above) display a significantly increased uptake of this LCM/ND lipid nanoemulsion version (as compared with the undetectable level of Emulsiphan nanoemulsion uptake in parenteral 3T3-L1 cells, which are noncancerous cells) (for added discussion, see Section 24.3 in ref. 1 ).
Besides the above dye-labeling experiments, both Constantinides et al 80 and Ho et al 81 have formulated LCM/ND lipid nanoemulsions with the anticancer drug, paclitaxel, and documented the successful delivery (intracellularly) of the carried drug to tumor cells of various types. 1
Of the above-mentioned “certain other (noncancerous) hyperproliferative-disease lesions/sites,” which overexpress scavenger receptors, one example is central nervous system (CNS) injury—that is, brain injury and/or spinal cord injury. Various published studies indicate increased scavenger receptor expression on “proliferating macrophages” and “activated astrocytes” arising after CNS injury. At the same time, this increased scavenger receptor expression, which probably mainly involves SR-BI (see Section 25.1.1 in ref. 1 ), provides a plausible avenue for targeted drug-delivery treatment of CNS-injury sites. Accordingly, Wakefield et al 82 examined the use of LCM/ND lipid nanoemulsion to deliver 7-hydroxycholesterol (7-OHC) to a radiofrequency (thermal) lesion in the rat brain. (7-OHC and other oxysterols have been reported, by other investigators, to inhibit astrogliosis both in vitro and in vivo (cf. 1 ). Wakefield et al 82 observed that the number of activated astrocytes were reduced when treated with 7-OHC delivered by the LCM/ND lipid nanoemulsion, while not affected by the same dose of intravenously injected 7-OHC in saline. It appears athat the mechanism of this enhanced delivery of 7-OHC to the brain-injury site, by a LCM/ND lipid nanoemulsion type, shares common features with the above tumor work (for added discussion, see Chapter 13 and Section 24.3 in ref. 1 ). The above interpretation of the data receives additional indirect support from published findings, of other investigators, which document the expression of SR-BI on astrocytes and vascular smooth muscle cells in adult mouse and human brains—as well as in Alzheimer’s disease brain. 1 Lastly, this documented ability of LCM/ND lipid nanoemulsion to function as a carrier of selected small molecular compounds would, of course, be potentially applicable to certain drug molecules already being used in research for treating Alzheimer’s disease and brain injury [see below].
Treating Brain Injury, Neuroinflammation, Oxidative Stress, and Alzheimer’s Disease via LCM/ND Nanoemulsions
The brief histological description above of brain-injury sites (cf. preceding Section) points to a larger pathophysiological overlap which exists between brain injury and Alzheimer’s-disease brain. First, as concerns brain injury, Wang et al 83 have pointed out that non-neuronal brain cells, especially astrocytes (the predominant cell type in the human brain), may exert an active role in the pathogenesis of traumatic brain injury (TBI). Activated astrocytes may contribute to increased oxidative stress and neuroinflammation following neurotrauma. Interestingly, the drug Edaravone has been used successfully, in past research, due to its neuroprotective and antioxidative effects on the brain after TBI. Wang et al 83 extended this research and found that, after intravenous administration (in rats), Edaravone treatment significantly decreased hippocampal neuron loss, reduced oxidative stress, and decreased neuronal programmed cell death as compared with control treatment. The protective effects of Edaravone treatment were also related to the pathology of TBI on non-neuronal cells, as Edaravone decreased both astrocyte and microglia activation following TBI. These authors conclude that the likely mechanism of Edaravone’s neuroprotective effect in the rat model of TBI is via inhibiting oxidative stress, leading to a decreased inflammatory response and decreased glial activation, and thereby reducing neuronal death and improving neurological function. 83 Similarly, Itoh et al 84 have reported that intravenous Edaravone administration (in rats), following TBI, inhibited free radical-induced neuronal degeneration and apoptotic cell death around the damaged area. Hence, Edaravone treatment improved cerebral dysfunction following TBI, suggesting its potential as an effective clinical therapy. 84
In view of the above description of TBI, the effects of the drug Edaravone, and the pathophysiological overlap of TBI with many characteristics of Alzheimer’s disease brain (cf. above), it is logical and consistent that Jiao et al 85 have recently reported that Edaravone can also ameliorate Alzheimer’s disease-type pathologies and cognitive deficits of a mouse model of Alzheimer’s disease. Specifically, besides reducing amyloid deposition and tau hyperphosphorylation, Edaravone was found to alleviate oxidative stress and, hence, attenuates the downstream pathologies including glial activation, neuroinflammation, neuronal loss, and synaptic dysfunction, and rescues the memory deficits of the mice. 85 (Note that Edaravone is a small-molecule drug, which is known to function as a free-radical scavenger; it currently is being used clinically in Japan to treat (acute ischemic) stroke patients. 83,85 ) Jiao et al 85 further state that their above findings suggest that Edaravone is a promising drug candidate for Alzheimer’s disease by targeting multiple key pathways of the disease pathogenesis. This recommendation by Jiao et al of Edaravone (for treating Alzheimer’s disease) fits well with the initial drug candidates suggested elsewhere, 76 based on low-molecular-weight and sufficient lipophilicity, for incorporation into the LCM/ND lipid nanoemulsion (as again proposed here) to treat Alzheimer’s disease. Since the Jiao et al recommendation is based in part on knowledge of failed clinical trials indicating that a single target or pathway does not work on this complex disease, 85 these investigators are understandably encouraged by a drug like Edaravone which targets multiple pathways of Alzheimer’s disease pathogenesis.
While the risk factors for dementia trigger widespread inflammation and oxidative stress (e.g., 86,87 ), it is also true that these 2 processes can result in more biological effects than enhanced calcium load in brain tissue and neurodegeneration (cf. 88 ). In fact, oxidative stress and inflammation each involve pathophysiological cascades associated with a wide range of pathologies and especially aging. However, these 2 processes/cascades are not always associated with biological damage. (For example, oxidative stress constitutes an important mechanism in many physiological processes, such as adaptations to physical exercise and cell signaling.) Yet, when oxidative stress and/or inflammation are dysregulated, their action is harmful. 88,89 (In this situation, one corresponding example [of many] occurs in Alzheimer’s disease, where growing evidence links the “reactive oxygen species” (ROS)-mediated damages with molecular targets including mitochondrial dynamics/function, autophagic pathways, and proteostasis balance. 90 ) Accordingly, Khalil et al 91 found that Alzheimer’s disease impaired the interaction of HDL (and ApoA-I) with the SR-BI receptor, and their experimental results indicated that such patients had higher levels of oxidative stress. 91,92 The authors concluded that their clinical study provides evidence for the first time that the functionality of HDL is impaired in Alzheimer’s disease, and that this alteration may be caused by Alzheimer’s disease-associated oxidative stress and inflammation. 91,92 This conclusion is consistent with earlier work where SR-BI was identified on astrocytes and vascular smooth muscle cells in Alzheimer’s disease brain, and has been demonstrated to mediate the adhesion of microglia to aggregated Aβ (cf. 92 ). Moreover, these authors further report that SR-BI mediates perivascular macophage response, and regulates Aβ-related pathology and cerebral amyloid angiopathy, in an Alzheimer’s-disease mouse model. 92
Covid-19, SAA, (“HDL-like”) LCM/ND Nanoemulsion, and (late-onset) Dementia
From Sect. 5 & 7 above, it is already apparent that brain injury can elicit a systemic inflammatory response medited through serum amyloid A (SAA) that contributes to the pathological outcomes (cf. 93 ). Also, from Sect 5, it is clear that SAA is a uremic toxin (and acute phase protein) which accumulates under inflammatory conditions associated with high cardiovascular morbidity and mortality in patients with sepsis; in addition, SAA is an alternate apolipoprotein of HDL (cf. 94 ). SAA accumulation turns HDL from an anti-inflammatory to a proinflammatory particle. In fact, as known from previous studies, high HDL-associated SAA concentration plays a major role in the “proinflammatory property” of these “(adulterated) HDL,” as shown by the artificially enriched SAA within HDL from healthy controls; this effect is comparable with HDL isolations from septic patients. In summary, SAA accumulation in HDL reduces its anti-inflammatory capacity by activating proinflammatory signaling pathways. SAA plays a key role in decreased HDL functionality and, therefore, represents an interesting therapeutic target for influencing the fate of cardiovascular-related disease. 94 Finally, as concerns this concept of an HDL-related therapeutic target, Sect. 5 also reviewed why “competitive binding” between SAA and (HDL-like) LCM/ND nanoemulsion particles, at (human)CLA-1/(rodent)SR-BI receptors, may well accompany clinical intravenous use of such nanoemulsion (which is repeatedly described in the literature as a targeted, apoA-I-based, drug-delivery agent 1 ) (cf. 95 ).
Meanwhile, much evidence indicates a protective (i.e., ordinarily anti-inflammatory) role for HDL, and its major apolipoprotein (apoA-I), in Alzheimer’s disease (e.g., 1,87,89,95 ) (cf. 96 ). Hence the above-proposed “competitive-binding,” between SAA and the LCM/ND nanoemulsion, could assist/enhance the “protective (ordinarily anti-inflammatory) role” of HDL—as well as provide targeted drug-delivery to the (human) brain cells bearing (CLA-1/)SR-BI receptors. This novel therapeutic-target approach has particular relevance to the current Covid-19 human pandemic. Namely, immune response and excessive inflammation in Covid-19 infection may accelerate the progression of brain inflammatory neurodegeneration, which plays a major role in Alzheimer’s disease pathology. In addition, individuals with type 2 diabetes are at increased risk for Alzheimer’s disease, as well as for severe outcomes after Covid-19 infection. 97 Severely affected Covid-19 cases experience high levels of proinflammatory cytokines and acute respiratory dysfunction. All these have the potential to cause cognitive decline (-- resulting from direct negative effects of the immune reaction, acceleration or aggravation of pre-existing cognitive deficits, and/or de novo induction of a neurodegenerative disease). Accordingly, patients surviving Covid-19 are at high risk for subsequent development of neurological disease and in particular Alzheimer’s disease. 98
Concluding Remarks
Microvascular endothelial dysfunction precedes, often by decades, the cognitive decline associated with Alzheimer’s disease. The cardiovascular risk factors for this disease trigger widespread inflammation and oxidative stress, both of which can lead to BBB disruption. Past studies (e.g., 41,99 ) have shown that low-grade inflammation and endothelial dysfunction contribute to reduced information processing speed and executive functioning in an older population. These interacting processes involve pathophysiological cascades which lead to neuronal (intracellular) Ca2+ increase, neurodegeneration, gradual cognitive/memory decline, and eventually dementia. By incorporating appropriate drug(s) into biomimetic (lipid cubic phase) nanocarriers, one obtains a multitasking combination therapeutic which targets certain cell-surface scavenger receptors, mainly class B type I (i.e., SR-BI), and crosses the BBB. Such biomedical application of colloidal drug-nanocarriers can potentially be extended to the treatment of complex medical disorders like dementia. 41,99 Recent reviews (e.g., 90,100,101 ) of human Alzheimer’s-disease studies have noted a significant elevation in inflammatory mediators in the cerebral microcirculation; crucially, inflammation has a key role in linking several types of vascular and neuronal damage (in Alzheimer’s-disease brain) to cardiovascular risk factors, such as arterial stiffness and hypertension. 90 In addition, various past studies indicate that inflammation also plays an important role in the process of Aβ deposition and, therefore, the inhibition of inflammatory cascades may attenuate amyloidogenic processes—such as Alzheimer’s disease. Hence, an effective preventive and therapeutic strategy could be based upon nanotargeting drug(s), using lipid nanocarriers, toward a major SAA receptor responsible for numerous SAA-mediated cell signaling events leading to cognitive decline and eventually Alzheimer’s disease or (late-onset) dementia. Lastly, it has been reconfirmed in the current literature that receptor-mediated endocytosis/transcytosis via lipoprotein receptors, particularly scavenger receptors including SR-BI, remains a major route for drug delivery across the blood–brain barrier; namely, recently published work has demonstrated that nanocomplexes can be readily transported into brain capillary endothelial cells (bovine and porcine) via SR-BI receptor-mediated endocytosis ( 102 ; see also 103 - 105 ). Accordingly, endothelial modulation and repair become feasible by pharmacological targeting 106 - 114 via SR-BI receptors (cf. 115 ). Moreover, the effects of the various cell types targeted (via SR-BI) may be additive, multiplicative, or otherwise synergistic. This therapeutic approach receives added impetus from continual findings of cerebrovascular pathology 116 - 122 and brain arterial aging 48,122 - 124 accompanying, and an apparent endothelium-dysfunction involvement 48,106 - 115,117 - 132 in, both Alzheimer’s disease and its major risk factors. 125 - 142 In addition, recent research (e.g., 143 - 146 ) repeatedly indicates for several age-related diseases, including cardiovascular and neurodegenerative disease, that accompanying the aging process there is often a decreased ability for fine control of systemic inflammation (i.e., the human immune system often dispalys a progressive and chronic tendency toward a proinflammatory phenotype, also called “inflamm-aging”) 143 (cf. 147 ). At the same time, the immune response and excessive inflammation (which is commonly observed in the very recent human coronavirus (Covid-19-) pandemic 148 - 153 ) may accelerate the progression of brain inflammatory neurodegeneration, 97,154,155 and the hippocampus is reported to be particularly vulnerable to coronavirus infections—which increases the probability of post-infection memory impairment and accelerating progression of Alzheimer’s disease. 155 Hence, the proposed multitasking combination therapeutic, using (biobased) LCM/ND nanoemulsion(s), may also display greater effectiveness at different stages of Alzheimer’s disease; as a result, this multitasking (drug-delivery) therapeutic could represent a promising way to treat, delay, or even prevent the disease in the future.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.S.D. is employed at Cav-Con Inc. The actual “LCM/ND nanoemulsion (nanoparticle)” described in this review is not a finished/manufactured product, and is not on the market for sale. Moreover, the LCM/ND nanoemulsion itself has only been employed in animal studies (i.e., has never been employed in a clinical study nor in any formal clinical trial).
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.