
Abstract
Alzheimer’s disease (AD) is the most common form of dementia in elderly individuals and is associated with progressive neurodegeneration of the human neocortex. Thiamine levels and the activity of thiamine-dependent enzymes are reduced in the brains and peripheral tissues of patients with AD. Genetic studies have provided the opportunity to determine what proteins link thiamine to AD pathology (ie, transketolase, apolipoprotein E, α-1-antitrypsin, pyruvate dehydrogenase complex, p53, glycogen synthetase kinase-3β, c-Fos gene, the Sp1 promoter gene, and the poly(ADP-ribosyl) polymerase-1 gene). We reviewed the association between histopathogenesis and neurotransmitters to understand the relationship between thiamine and AD pathology. Oral thiamine trials have been shown to improve the cognitive function of patients with AD; however, absorption of thiamine is poor in elderly individuals. In the early stage of thiamine-deficient encephalopathy (Wernicke’s encephalopathy), however, parental thiamine has been used successfully. Therefore, further studies are needed to determine the benefits of using parental thiamine as a treatment for AD.
Introduction
Glucose is required for energy generation in the brain. Nearly 30% of brain glucose undergoes complete oxidation through the mitochondrial tricarboxylic acid cycle. 1 Thiamine is transported to the brain and is phosphorylated to thiamine diphosphate (TDP) by thiamine pyrophosphorylase. In the neuronal metabolism of glucose, TDP is an essential coenzyme for mitochondrial pyruvate, α-ketoglutarate dehydrogenases (KGD) complexes, and cytosolic transketolase (TK). 2,3 In thiamine deficiency, the levels of thiamine-dependent and non-thiamine-dependent enzymes (succinate and malate dehydrogenase) in the tricarboxylic acid cycle are reduced in the mouse brain. 4 Similarly, the activity of brain KGD is also reported to decrease in Alzheimer’s disease (AD), 5 suggesting that thiamine may have a role in the pathogenesis of AD. Therefore, we review the role of thiamine in AD pathology.
The Relationship Between Thiamine and AD
Nutritional Factors
Disturbance of glucose metabolism is a prominent characteristic in the brains of patients with AD, and type 2 diabetes mellitus has been identified as a risk factor for AD. 6 -8 Low blood thiamine levels, erythrocyte TK activity, and high erythrocyte thiamine pyrophosphate (TPP) activity have been documented in diabetic patients. 9,10 In addition, plasma thiamine level has been shown to be decreased by 76% in patients with type 1 and 75% in patients with type 2 diabetes and associated with increased renal clearance and fractional excretion of thiamine. 11 Similarly, low plasma thiamine levels have also been reported in patients with AD. 12 -14 In AD brains, the activities of the KGD were reduced by more than 75% and those of TK by more than 45%; significant abnormalities of TK were identified in red blood cells and cultured fibroblasts of patients with AD. 15 These findings suggested that thiamine may have a role in the pathogenesis of AD.
Genetic Factors
Genetic studies provide an excellent opportunity to link molecular variations with epidemiological data. DNA sequences variations such as polymorphisms have modest and subtle biological effects. Receptors play a crucial role in the regulation of cellular function, and small changes in their structure can influence intracellular signal transduction pathways.
Transketolase has been used to assess thiamine activity in mammalian tissue. Structural abnormalities of TK occur in a high proportion of patients with AD. 16,17 Fibroblasts from patients with AD propagated at different pH values, within a range of 7.3 to 7.8, have been shown to exhibit TK abnormalities. 18 In addition, proteolytic cleavage has been shown to decrease TK activity 19 and might be used as biochemical marker in fibroblasts of patients with AD. Similarly, TK variants have been found in patients with thiamine-deficient encephalopathy. 20 The TK extracted from the fibroblasts of thiamine-deficient alcoholic patients and their sons has been shown to have a decreased affinity for TPP. 21,22 Finally, Heap et al 23 investigated the affinity of erythrocyte TK for its cofactor in alcoholics with cognitive impairment. They reported that, despite adequate nutrition, this patient subgroup has altered TK-binding. These reports, however, did not rule out the alcoholic effect on the TK.
Apolipoprotein E (ApoE) is a major genetic factor identified in AD. Carriers of at least 1 ApoE ∊4 allele have an increased risk of developing AD. 24,25 ApoE has been shown to be significantly altered in the cerebrospinal fluid (CSF) of patients with AD. 26 In addition, capillary cerebral amyloid angiopathy has been identified as a distinct ApoE ∊4-associated subtype of sporadic AD, 27 which may determine the clinical phenotype of AD. 28 Patients expressing this ApoE genotype are known to have significant impairment in memory retention. Conversely, ApoE was not found to be a risk or a protective factor for AD in an Ecuadorian population. 29 On the other hand, Muramatsu et al 30 reported that the frequency of the ApoE ∊4 allele was significantly higher in patients with thiamine-deficient dementia.
α-1-Antitrypsin (ATT) has been identified as a potential biomarker in the plasma of patients with AD. 31 Higher levels of ATT in CSF were associated with the clinical diagnosis of AD. 26,32 In a study consisting of 1136 patients presenting with cognitive disorders, polymorphisms in ApoE, the gene for hemochromatosis and ATT were present in up to 40% of the patients. 33 Similarly, ATT polymorphism is associated with increased incidence of thiamine deficiency. 34
Genetic defects in pyruvate dehydrogenase complex (PDHC) are known to cause lactic acidosis, neurological deficits, and premature death. 35 Patients with these defects show reduced activity of PDHC and pyruvate dehydrogenase (PDH) E1-alpha subunit and decreased affinity of PDHC for TPP. 36,37 Thiamine treatment is very effective in some patients with PDHC-deficiency. 37,38 Thiamine regulates the expression of enzymes that require thiamine as a cofactor and thiamine deficiency has been shown to reduce the messenger RNA (mRNA) levels of TK and PDH. 39
There are numerous potential gene products that are transcriptionally activated by p53 and are involved in cell cycle arrest or apoptosis. 40 In cellular models of AD, p53 has been found to be undergoing conformational changes that make cells less vulnerable to stressors or genotoxic insults. 41,42 DNA damage has been shown to increase p53 DNA-binding activity. Furthermore, TDP has been shown to inhibit p53 binding, and thiamine has been shown to inhibit intracellular p53 activity. 43 Recently, p53 and glycogen synthetase kinase-3β (GSK3β) have been linked to AD. 44,45 The GSK3β is a protein kinase involved in many physiological processes, for example, cell structure, metabolism, gene expression, and apoptosis. In addition, the interaction of p53 and GSK3β has been shown to lead to an increased activity of both proteins. Exposure to pyrithiamine, an anti-thiamine compound, has also been shown to increase β-amyloid protein accumulation and GSK3 activity in the brain. 46 In an animal AD model, benfotiamine was found to improve the cognitive function, reduce amyloid deposit, and suppress GSK3 activity. 47 These findings suggested a role of thiamine in controlling the activity of p53 in patients with AD.
The proto-oncogene protein c-Fos codes for a nuclear protein involved in growth-related transcriptional control. Activation of c-Fos has been suggested to contribute to β-amyloid protein-induced neurotoxicity in AD, 48 and over-expression of c-Fos mRNA has been reported in AD. 49,50 Similarly, c-Fos expression has been shown to increase with the progression of thiamine deficiency and has been associated with neuronal loss in the thalamus and inferior colliculus of the thiamine-deficient rat. 51 -53
Thiamine uptake in the human intestine occurs via a specialized carrier-mediated mechanism. The human thiamine transporters were found to be expressed in the human intestine, which are regulated via Sp1 promoter elements. 54 Sp1 transcription factor may be involved in regulating the expression of several AD-related proteins. Abnormal Sp1 transcription factor has been reported in AD. 55,56 The regulatory region of the amyloid precursor protein (APP) gene contains sites recognized by the Sp1 transcription factor, which has been shown to be required for the regulation of APP and Aβ. 57 BACE1, the major β-secretase involved in cleaving APP promoter contains a functional Sp1 response element, and overexpression of the Sp1 transcription factor potentiates BACE gene expression and APP processing to generate Aβ. 58 Sp1 and signaling mother against decapentaplegic peptide (Smad) transcription factors cooperate to potentiate transforming growth factor-β-dependent activation of APP. 59 These findings suggested a role of Sp1, the thiamine homeostasis, in AD.
Poly(ADP-ribosyl) polymerase (PARP) is a nuclear protein that contributes to both neuronal death and survival under stress condition. Poly(ADP-ribosyl) polymerase cleavage is enhanced in peripheral blood mononuclear cells from patients with mild cognitive impairment. 60 Enhanced-PARP activity is reported in AD and is suggested as a marker for AD. 61,62 Poly(ADP-ribosyl) polymerase polymers increased with age in the brains of Alzheimer mouse model and β-amyloid-activated PARP polymers inducing astrocytic metabolic failure and neuronal death in response to oxidative stress. 63 Poly(ADP-ribosyl) polymerase polymorphism is shown to modify the risk of AD in both an independent manner and interaction with proinflammatory interleukin-1A gene. 64 Poly(ADP-ribosyl) polymerase gene is highly associated with AD susceptibility. The distributions of PARP haplotype, Ht3-TT, and Ht4-CC were significantly associated with an increased risk of AD, whereas the Ht1-TC haplotype showed a protective effect for AD when compared with the control participants. 65 Conversely, thiamine has a cytoprotective effect on cultured neonatal rat cardiomyocytes under hypoxic insult by inhibiting PARP cleavage and DNA fragmentation. 66 Furthermore, benfotiamine prevents liposaccharide-induced apoptosis and PARP cleavage in mouse macrophage cell lines. 67 The relationship between thiamine and AD has been summarized in Table 1.
The Relationship Between Thiamine and Alzheimer’s Disease

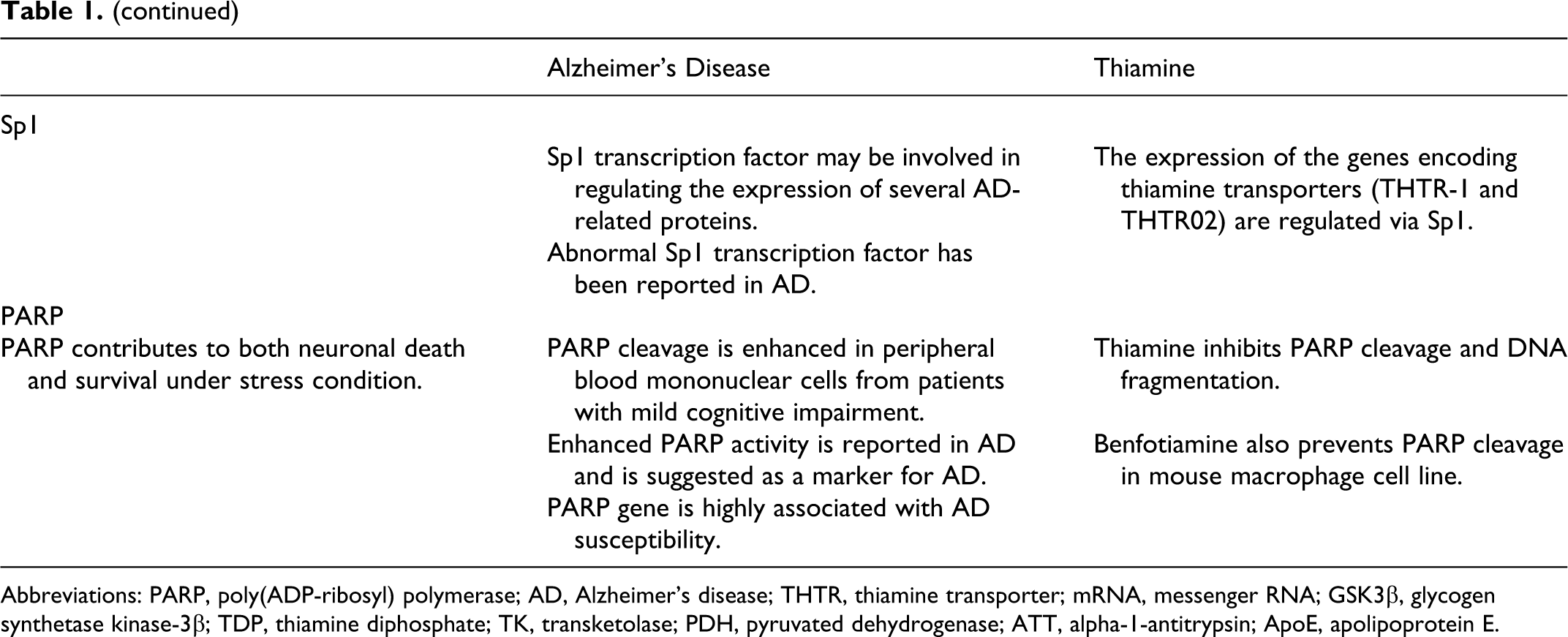
Abbreviations: PARP, poly(ADP-ribosyl) polymerase; AD, Alzheimer’s disease; THTR, thiamine transporter; mRNA, messenger RNA; GSK3β, glycogen synthetase kinase-3β; TDP, thiamine diphosphate; TK, transketolase; PDH, pyruvated dehydrogenase; ATT, alpha-1-antitrypsin; ApoE, apolipoprotein E.
Histopathogenesis
The pathogenesis of AD is complex. Several factors, including amyloid plaques, neurofibrillary tangles, the loss of neuronal cells, and inflammatory processes are likely to contribute to the development of the disease.
Amyloid Plaques and Neurofibrillary Tangles
The accumulation of β-amyloid protein is considered to be a main cause of neuronal cell death in AD. Amyloid precursor protein protects against excito-toxicity following neuronal injuries by regulating the function of the glial glutamate transporters. Prolonged treatment with APP has been shown to augment glutamate clearance by cultured astrocytes and induce a dramatic decrease in glutamatergic synaptic activity of neurons cocultured with astrocytes. 68 In postmortem tissue, neurofibrillary tangles have been found in the nucleus basalis of chronic alcoholics. 69 Thiamine deficiency in mice led to the accumulation of APP within neuritic clusters in damaged brain regions. 70,71 Abnormal clusters were found to occur only in areas of neuronal damage, for example, the thalamus, mammillary bodies, and inferior colliculus. These clusters appeared as either irregular clumps or round or oval rosettes that resembled the neuritic component of the amyloid plaques seen in AD. 71
Loss of Neuronal Cells
In patients with AD, β-amyloid activates macrophages and microglia, producing inflammation that accelerates neuronal damage. 72 On the other hand, thiamine-deficient encephalopathy is characterized by a selective loss of neurons in the midbrain, thalamus, and cerebellum. 73 In addition, thiamine deficiency also produced fiber cell degeneration in mouse lenses. 74
Inflammatory Processes
It is widely accepted that microglial-mediated inflammation contributes to the progression of AD. 72 Similarly, microglial activation has been shown to occur in the early and late stages of thiamine-deficient animal models. 75 -78
Neurotransmitters
Acetylcholine (ACh) and norepinephrine (NorEpi) are the most common neurotransmitters associated with the pathophysiological conditions observed in AD. It is hypothesized that these neurotransmitters are hypoactive in AD.
Acetylcholine
Regions throughout the neocortex receive cholinergic inputs from the basal forebrain. Neurotransmission mediated by ACh has been shown to play an important role in attention processing. 79 Moreover, cortical deficiencies in cholinergic neurotransmission are known to contribute to the characteristic cognitive deficits associated with AD. 79 It has been shown that choline acetyltransferase activity, synaptic reuptake of choline, and ACh synthesis are reduced in AD. 80,81 Alterations in the density of cholinergic and noncholinergic receptors for glutamate, noradrenaline, and serotonin have been reported in transgenic Tg2576 mice with β-amyloid plaque pathology. 82 On the other hand, it has been suggested that thiamine-deficient encephalopathy involves impairment in cholinergic neurotransmitter function. Thiamine is an acoenzyme required for the synthesis of ACh, which is the neurotransmitter that is most commonly deficient in AD. In the neocortex, impaired coupling of muscarinic M1 receptors to G-proteins has been shown to be associated with the severity of dementia in AD. 83 Moreover, it has been shown that the synthesis of ACh is impaired in the brains of thiamine-deficient rats, 84 leading to a significant reduction of neuronal ACh levels. 85 Animal studies have also suggested that thiamine is involved in the presynaptic release of ACh; it has been shown that thiamine binds to nicotinic receptors and may exhibit anticholinesterase activity. 86 In addition, thiamine deficiency has been shown to induce an early central muscarinic cholinergic lesion. 87
Norepinephrine
The locus coeruleus (LC) is the major source of NorEpi, which is known to reduce neuroinflammation in the brain. Decreases in the number of NorEpi-containing neurons in the LC suggests that there should be reduced NorEpi activity in patients with AD. 88 Similarly, the concentration of NorEpi within CSF of participants with thiamine-deficient encephalopathy are also significantly reduced. 89 Using biochemical analyses, Mair et al 90 demonstrated that the concentration of NorEpi was significantly reduced in the brain (cortex, hippocampus, and olfactory bulb) accompanied by a concomitant decrease in behavioral measures of learning and memory in thiamine-deficient rats, suggesting that NorEpi deficits may contribute to the memory impairment. Moreover, Mousseau et al 91 demonstrated a brain region-selective vesicular dysfunction in catecholamine metabolism in an experimental thiamine deficiency model and suggested that it may account for certain neurobehavioral effects commonly encountered in thiamine deficiency. Furthermore, thiamine and its derivative (allylthiamindisulfide) significantly increased the plasma concentrations of noradrenaline and adrenaline in rats. 92
Glutamate
Glutamate is synthesized from glucose and glutamine in presynaptic neuronal terminals and serves as a major excitatory neurotransmitter in the central nervous system (CNS). The capacity for cognition and memory is derived from various input and output pathways between the hippocampus and neocortex that rely on glutamatergic signaling. 93 Anomalies in glutamate homeostasis may contribute to the pathological processes involved in AD. Uptake by the vesicular glutamate transporter has been shown to be decreased in patients with AD. 94 Glutamate transporters are believed to protect neurons against excito-toxicity by removing extracellular glutamate, and the β-amyloid protein prevents excito-toxicity via the recruitment of glial glutamate transporters. 95 In transgenic models of early-stage amyloid pathology, a significant reduction in the density of cholinergic, glutamatergic, and GABAergic presynaptic boutons has been observed. 96 In addition, alterations in the expression of glutamatergic transporters and receptors have been reported in cases of sporadic AD. 97 Studies have shown that decreased function of glutamate transporters in AD might lead to neurodegeneration. The excitatory amino acid transporter-2 (EAAT-2), a glutamate transporter, is expressed in astrocytes. In AD, many EAAT-2-positive neurons have been shown to have abnormalities in the cytoskeleton and the tau-protein. Moreover, these neurons were often found to have condensed and shrunken nuclei. 98 Neuronal glutamate transporter and EAAT-2 mRNA splice variants have been found in the brain of patients with AD. 99,100 In addition, a significant reduction in EAAT2 protein expression levels has been reported in the mid-frontal cortex of patients with AD. 101 Glial EAAT-1 was shown to be selectively expressed in degenerating neurons and dystrophic neuritis in AD. 102 Altered glutamate transport and aberrant EAAT-1 expression were found to be decreased in patients with AD brains. 103,104 On the other hand, glutamate transporters have been shown to be downregulated in thiamine-deficient astrocytes. 105,106 It has also been shown that the levels of EAAT-1 and EAAT-2 are diminished by 62% and 71%, respectively, in individuals with thiamine-deficient encephalopathy and treatment with N-acetylcysteine also prevented the downregulation of EAAT-2 in the medial thalamus and ameliorated the loss of several other astrocytic proteins. 107 In addition, treatment with pyrithiamine, a central thiamine antagonist, has been shown to decrease the protein levels of astrocytic glutamate transporters in the medial thalamus. 108
Histamine
Histamine (HA) has a wide spectrum of biological actions in the CNS. Increased levels of HA have been noted in the brain, serum, and CSF of patients with AD. 109,110 Other studies, however, have shown a decrease in HA content in the hypothalamus, hippocampus, and temporal cortex of AD brains. 111,112 In severe cases of AD, an increased density of H3 HA-receptors has been detected in the medial temporal cortex. 112 The novel H3 HA-receptor antagonist (GSK189254) has been shown to increase the release of ACh, noradrenaline, and dopamine in the anterior cingulate cortex and ACh in the dorsal hippocampus. In addition, GSK189354 was found to significantly improve the performance of rats in a number of cognition testing paradigms. 113 Similarly, increased HA release was observed in the lateral thalamus in thiamine-deficient rats. 114 These rats demonstrated mouse-killing aggression (muricide). In addition, H1 HA-receptor antagonists (diphenhydramine, promethazine, and chlorpheniramine) have been shown to suppress muricide. 115 In thiamine-deficient rats, HA levels were found to be lower in the hippocampus, amygdala, olfactory bulb, thalamus, and pons-medulla; but, higher HA levels were observed in the hypothalamus. 116 After dietary thiamine supplements, however, these HA levels returned to normal in these rats. The relationship between thiamine and AD on the neurotransmitter has been summarized in Table 2.
Thiamine and Neurotransmitters
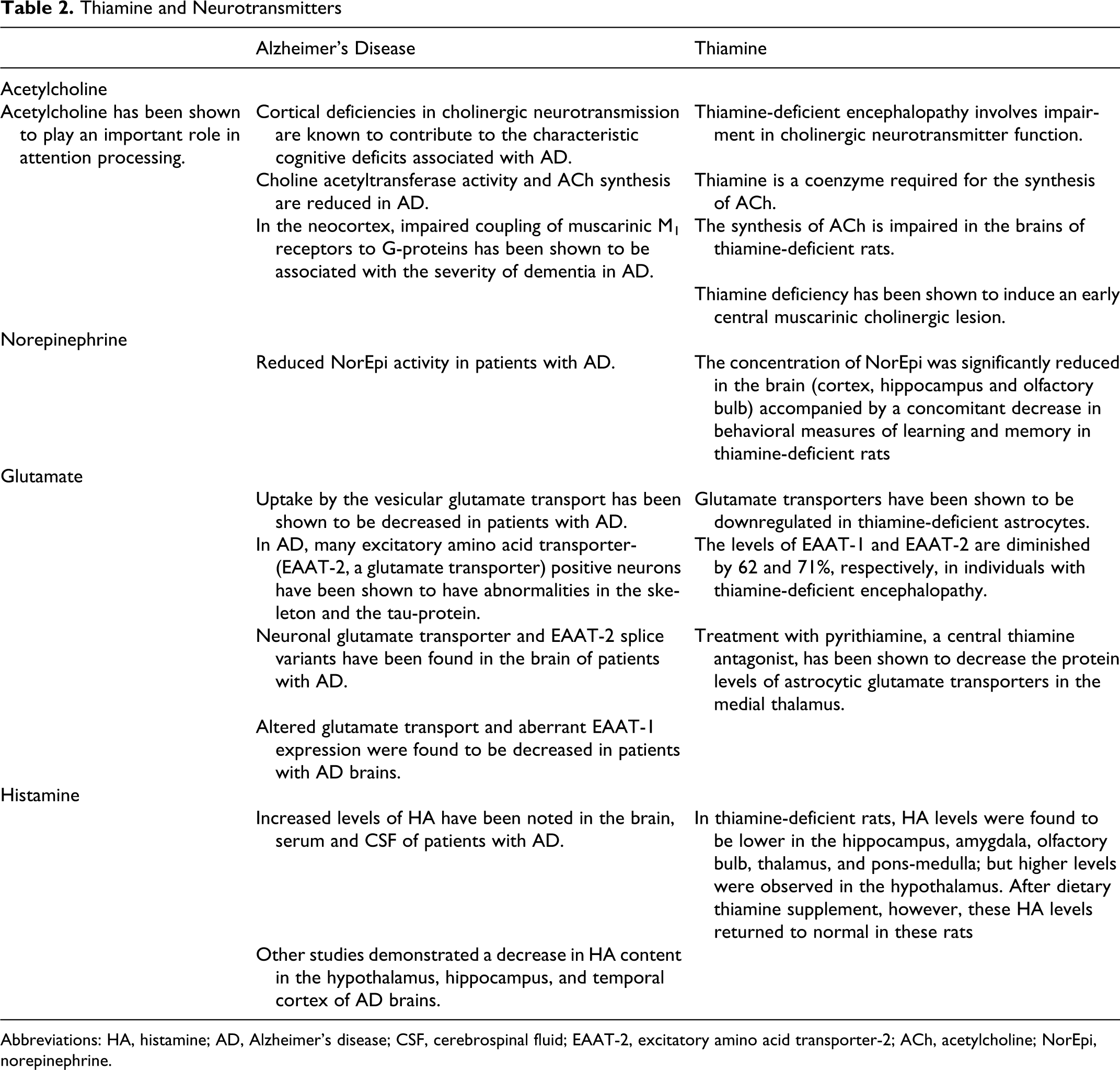
Abbreviations: HA, histamine; AD, Alzheimer’s disease; CSF, cerebrospinal fluid; EAAT-2, excitatory amino acid transporter-2; ACh, acetylcholine; NorEpi, norepinephrine.
Role of Thiamine in AD
Thiamine Trials
In patients with AD, fursultiamine, a derivative of thiamine, was found to have a mild beneficial effect using an oral dose of 100 mg/d in a 12-week trial.
117
In a short-term trial using 3 g/d of oral thiamine, Blass et al
118
demonstrated that the global cognitive rating by the Mini-Mental State Examination was improved in patients with AD. In another study using 3 to 8 g/d of oral thiamine, Meador et al
119
reported a mild beneficial effect of thiamine in patients with AD. The long-term oral administration of thiamine at 3 g/d, however, did not slow the progression of AD.
120
Eight weeks benfotiamine treatment was found to improve the cognitive function and reduce both number of amyloid plaques and phosphorylated tau levels via a thiamine-independent mechanism in an animal model of AD.
121
These effects, however, were not found using fursultiamine. Interestingly, benfotiamine was unable to raise the levels of intracerebral thiamine phosphate derivatives.
122
In addition, it was found that benfotiamine does not easily cross neuroblastoma cell membrane in cultured cells.
123
Sulbutiamine, a lipid-soluble thiamine disulfide derivative, that increases thiamine derivatives in the brain and in cultured cells, can be used as a CNS drug. In rats, chronic treatment with sulbutiamine was shown to improve memory in an object recognition task and reduce the amnesic effects of dizocipine, which blocks the N-methyl-
Pharmacokinetics
The intestinal absorption of thiamine is sufficient in young people, but may be reduced with age. 126 Schaller and Holler 127 reported that intestinal alkaline phosphatase (ALP) is involved in the active thiamine absorption in the intestinal tract. Furthermore, Rindi et al 128 found that intestinal ALP can transphosphorylate thiamine to thiamine monophosphate during intestinal transport in rats. Without ALP, it was found that thiamine could not be transported into the lumen of the gastrointestinal tract. 129 In addition, it has been shown that the intestinal ALP activity declines significantly with age in mice. 130 Enzymatic activity of ALP in the duodenum was also found significantly higher in 5-month-old rats compared with other age groups, especially 2.5 weeks and 23 months old rats. 131 The decrease in the intestinal ALP activity in old rats has been attributed to the reduction in the number of enterocytes caused by the age-induced atrophy of the intestinal mucosa. 132 In addition, oral absorption of thiamine was found to be poor in older individuals. 133 In humans, single oral doses of thiamine higher than 2.5 to 5 mg are largely unabsorbed. 134,135 Nichols and Basu 136 reported that almost half of the elderly individuals (older than or equal to 65 years) had an effect of TPP on the TK activity above 14%, which is suggestive of thiamine-deficient state. It should be noted, however, that the daily thiamine intake of these patients was above the recommended requirement (>0.4 mg/1000 kcal). Baker et al 137 demonstrated that thiamine deficiency in individuals older than 60 years could only be corrected by intramuscular administration of thiamine. Sasaki et al 138 reported a case of thiamine deficiency with psychotic symptoms. The patient’s condition was ameliorated only by repeated administrations of intravenous thiamine.
Conclusion
The relationship between thiamine and AD has been discussed, but it could not rule out the mass effect changes from cerebral atrophy in AD. Thiamine levels and activity of thiamine-dependent enzymes are reduced in the brains and peripheral tissues of patients with AD. Genetic studies, however, have provided the opportunity to determine what proteins link thiamine to AD pathology. Although oral thiamine supplement have been shown to improve cognitive function in patients with AD; but, thiamine absorption decreases with advancing age. In the early stages of thiamine-deficient encephalopathy (Wernicke’s encephalopathy), patients were found to respond rapidly to large doses of parental thiamine. The initial dose of thiamine is usually at 100 mg 2 to 3 times daily for 1 to 2 weeks. Therefore, further studies to determine the therapeutic value of parental thiamine for treating AD would be of great interest.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.