
Abstract
The pathogenesis of Alzheimer’s disease involves multiple pathways that, at the macrolevel, include decreased proliferation plus increased loss affecting neurons, astrocytes, and capillaries and, at the subcellular level, involve several elements: amyloid/amyloid precursor protein, presenilins, the unfolded protein response, the ubiquitin/proteasome system, the Wnt/catenin system, the Notch signaling system, mitochondria, mitophagy, calcium, and tau. Data presented show the intimate, anatomical interactions between neurons, astrocytes, and capillaries; the interactions between the several subcellular factors affecting those cells; and the treatments that are currently available and that might correct dysfunctions in the subcellular factors. Available treatments include lithium, valproate, pioglitazone, erythropoietin, and prazosin. Since the subcellular pathogenesis involves multiple interacting elements, combination treatment would be more effective than administration of a single drug directed at only 1 element. The overall purpose of this presentation is to describe the pathogenesis in detail and to explain the proposed treatments.
Introduction
The pathogenesis of Alzheimer’s disease (AD) incorporates many different elements and processes. At the macrolevel, the cellular disturbance that is the basis of AD involves decreased proliferation of stem/progenitor cells plus increased loss affecting neurons, astrocytes, and capillaries, causing impairment of progression into various stages of cellular maturity. The net ultimate result that leads to AD is less renewal of mature cells than is required to compensate for cellular loss. The consequent, impaired functions of neurons, astrocytes, and capillaries cause brain dysfunction and the dementia. At the subcellular level, the several pathogenetic mechanisms that induce the macrocellular abnormalities include amyloid and its precursors, presenilins (PS), the unfolded protein response (UPR), the ubiquitin/proteasome system (UPS), the Wnt/catenin system, the Notch signaling system, mitochondria, mitophagy, calcium, and tau. The importance of recognizing the early roles in the pathogenesis of these mechanisms is that rational therapy to prevent the macrocellular dysfunctions causing AD depends upon reversing their impairments. Although almost all research on therapy have focused on affecting just one of the subcellular elements, a more overarching yet challenging approach is to consider the entire web of interactions.
First, in part 1, data are presented about the intimate and critically important, anatomical interactions between neurons, astrocytes, and capillaries; this is followed by data showing the blocks to renewal of those cells that occur in normal aging but are amplified in AD. Part 2 describes the interacting, subcellular mechanisms that affect neurons, astrocytes, and capillaries. Part 3 describes the interactions between the subcellular mechanisms. Finally, part 4 proposes treatments that are currently available and that might correct dysfunctions in the subcellular factors that are the drivers of AD.
Although the results and conclusions of the studies supporting the data presented here may sometimes conflict with those from other studies, the data cited represent what seems to be the majority opinion concerning the particular topic.
Part 1: The Macrocellular Pathogenesis
Some Details of the Anatomy and Physiology of the Cellular Participants
Neural stem cells (NSCs) persist in the adult brain which, like all other organs, undergoes constant cell loss and cell renewal. Cell renewal in the brain largely happens in the anterior part of the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the hippocampal dentate gyrus.
Adult neurogenesis consists of 4 overlapping stages: (1) proliferation in the SVZ and SGZ of stem/progenitor cells, (2) differentiation into immature neuroblasts, (3) maturation of neurons, and (4) migration into neuronal networks. Interruption of the process, which may occur at any of the stages, is evaluated by examining the expression of various neuronal surface proteins. Proliferation, which putatively identifies stem/progenitor cells, is identified by the surface expression of many proteins including Ki67, Sox-2, Tbr2, MCM2, TUC-4, and doublecortin (DCX); Musashi-1 is a neural RNA-binding protein enriched in NSCs; immature neurons also express DCX and bromodeoxyuridine (BrdU, which labels newly synthesized DNA and identifies newly generated cells); mature neurons express NeuN.
The early pathogenesis of AD affects neurons, astrocytes, endothelial cells, and capillaries, all to an approximately equal degree, so it is noteworthy that the cell renewal process in the SVZ and SGZ involves each of those cell types. 1,2 Astrocytes direct stem cells toward neurogenesis 3 and may act as NSCs, 4 and endothelial cells also participate in regulating proliferation of neural precursors. 5
It is important to consider neurons, astrocytes, and capillaries as a unit because of their close anatomical relationship. The interactions between neurons and astrocytes are intimate, underlie brain function, and are affected early in AD. Protoplasmic astrocytes are the main sort in the gray matter of the brain. They have 5 to 10 main stem branches from each of which arise many finely branching processes that contact several hundred dendrites from multiple neurons and synapses. 6 In this way, each astrocyte supports and modulates the functions of roughly 2 million synapses. 7 The astrocytes’ processes envelop all synapses, where they play essential roles in transmitter homeostasis by clearing the neurotransmitters (glutamate, GABA, etc) from the synaptic space. Astrocytes have the same neurotransmitter receptors and ion channels as have neurons (for a comprehensive review, see the study by Verkhratsky et al 7 ).
Aside from cross talk between the 3 cell types, both neurons and astrocytes affect capillaries by secreting the angiogenic factors, fibroblast growth factor 2, and vascular endothelial growth factor. 8 Another angiogenic factor released by astrocytes is the high-mobility group box 1, which ligates its receptor on endothelial cells and induces proliferation. 9 Endothelial cells also participate in regulating the proliferation of neural precursors. 10
Astrocytes are central to the homeostasis of electrolytes and neurotransmitters in the brain. High extracellular concentrations of K+ ions from neural activity enter astrocytes, together with water via channels activated by aquaporins. Aquaporin-4 localizes to the end-feet of astrocyte processes surrounding capillaries; it regulates water permeability, thus transport of drugs through the blood–brain barrier (BBB). Aquaporin-4 knockout mice showed about 60% less coverage of cerebral capillaries by astrocytic processes and decreased efficacy of fluoxetine 11 ; the mice also had cognitive defects. 12 A small study showed cognitive improvements in patients with mild cognitive impairment (MCI) receiving fluoxetine as compared with controls receiving placebo. 13 Extracellular glutamate released by neurotransmission is taken up by perisynaptic astrocytes, utilizing specific glutamate transporters and preventing excitotoxicity. Glutamate transport is accompanied by influx of Na+ and H+ and efflux of K+. The increased Na+ stimulates glycolysis. Glutamate in the astrocytes is converted to nontoxic glutamine by glutamine synthetase, transported to the presynaptic space, and from there into neuronal cytoplasm, where it is reconverted into glutamate. Signaling is accomplished by the so-called tripartite synapse that is formed by pre- and postsynaptic components plus astrocytic presynaptic processes. Importantly, glutamate transport is decreased in AD brain. 14
Capillaries are formed by endothelial cells, pericytes, and the capillary basal lamina onto which astrocytic feet are attached. Tight gap junctions join the endothelial cells and pericytes which, together with the basement membrane and processes of protoplasmic astrocytes that plaster the walls of neighboring capillaries, help form the BBB. Pericytes are contractile and regulate capillary diameter and blood flow. 15 It is important that brain endothelial cells differ from other endothelial cells, in that although they are lined by a fused basement membrane that is formed by the endothelial and astrocytic basement membranes, there is no large extracellular space between the brain microcapillaries and the neural compartment. 16 In cognitively normal individuals aged 23 to 91 years, a high-resolution method of dynamic contrast-enhanced magnetic resonance imaging (MRI) was used to assess permeability of the BBB in the living human brain based on the blood-to-brain transfer of gadolinium; it showed an age-dependent BBB breakdown in the CA1 and dentate gyrus regions of the hippocampus. 17
Changes in Cell Genesis Affecting Neurons, Astrocytes, and Capillaries May Occur With Normal Aging
Advanced age is a major risk factor for AD. In the normal brain, there is less cell renewal with increasing age, affecting neurons, astrocytes, and capillaries, which if excessive might lead to AD.
Several studies show that in the normal brain, the “graying of age” is a downstream block in the process of cell renewal, whereas in AD, that block occurs to a far greater degree. In fact, there is neuronal loss with normal aging of the brain. In aged controls, the CA1 area of the hippocampus showed a 67% neuronal loss and the subiculum a 32% loss. 18 Elderly persons without clinical or histopathological evidence of neuropathology had decreasing CA1 number with increasing age. 19 The dentate gyri from 51 humans aged from 1 day to 100 years, who had no clinical or histopathological evidence of neuropathology, showed proliferating neuroblasts and both immature and mature neurons, but the total numbers of cells with these markers decreased exponentially with increasing age. 20 Others confirmed that the number of NSCs in the SGZ remained stable during aging, 21 but the expression of a mature neuronal marker was delayed and early dendritic growth retarded. 22
Although the number of astrocytes, in human brains, was 19% lower in older than in the younger patients (P = .2, nonsignificant), it did not change much beyond age 65. 23 Nevertheless, there are both structural and physiological changes in astrocytes with aging. Glutamine synthase (GS), expressed solely in astrocytes, is required for the maintenance of synaptic transmission mediated by glutamate and GABA. Astroglial GS converts glutamate taken up by astrocytes to glutamine, which is then transported to neurons where it is reconverted to glutamate and to GABA. Astrocytes of older mice showed a decrease in GS levels. 24 Nestin (a marker of NSCs) and DCX (a marker of neuroblasts) were both reduced in the aging SVZ. 25 Aging astrocytes were more dispersed and had decreased numbers of cristae. 25 Parenthetically, it should be pointed out that results from rodent studies of astrocytes may not be fully transposable to humans because rodents’ astrocytes connect with <10% of the synapses than do humans’ astrocytes and that humans’ astrocytes have 16.5 times greater volume and they signal 5 times faster via calcium waves. 26
Other important changes in cerebral capillaries with advancing age include increases in the cross-sectional area of the capillary basement membrane, in the fraction of endothelial cell and pericyte cytoplasmic areas occupied by mitochondria, and in the size of the pericyte mitochondria, as well as a decreased capillary lumen area in the hippocampus. 27
The studies reviewed in the following sections provide details of the blocks that are found at every stage of the renewal process in neurons, astrocytes, and cerebral capillaries, of AD.
Blocks to Neurogenesis in AD Are Beyond Those in Normal Aging
Neuronal loss in the CA1 area of the hippocampus and the subiculum in AD was 23% more than in controls. 18 The decline of neurogenesis within the SVZ correlated with cognitive impairments in AD. 2 Whereas telomere shortening occurs normally with cellular senescence, telomeres in hippocampal neurons in AD were 23% shorter than in controls, suggesting premature neuronal senescence and death of neurons. 28 And cognitively normal elders showed more subsequent development of AD when telomeres at baseline were shorter rather than longer. 29
Decreased proliferation and survival of stem/progenitor cells
Proliferation of NSC in the SVZ was reduced by 3.5-fold in 2-year-old wild-type (WT) mice as compared with 2-month-old ones; the secondary neurospheres generated per subcloned primary neurosphere also declined significantly in older mice. 30 Between 4 and 12 to 24 months of age in WT rats, the number of proliferating Ki67+ cells declined by 0.9-fold, 21 but in AD brains, the decline was 9-fold in Musashi-1-labeled neural progenitor cells (NPC) in the SVZ. 31 Mice transgenic for the familial AD (FAD)–linked mutant APPswe/PSIΔE9 showed severely impaired proliferation of NPC in the SVZ and subgranular layer as early as 2 months of age and, perhaps importantly, preceding amyloid deposition. 32 Other mice transgenic for FAD PS1 had impaired survival of NPC. 33
Decreased differentiation into immature neuroblasts and/or their decreased maturity into neurons
In both normal humans and animals, maturation is increasingly blocked with advancing age, but the block is amplified in patients with MCI or AD. For example, a group of patients with the mildest degree of clinically detectable AD had 32% fewer neurons in the entorhinal cortex (EC) than controls; neurogenesis in their dentate gyri did not proceed to completion; and these decrements in layers 2 and 4 were 90% and 70%, respectively, in those with severe dementia as compared with controls. 34 Neurons were reduced in the superior temporal sulcus by 53% compared with control brains, ranging from no loss in patients with symptoms <1 year to 75% loss in those with more advanced AD. 35 Decreases in individual laminae of the EC were also dramatic, with the number of neurons in layer 2 decreasing by 60% and in CA4 by 40% compared with controls. In that study, neuronal number in the EC was not related significantly to diffuse amyloid plaques or to total plaques. 34 In AD, the expressions of MAP 2a and 2b, which label mature neurons, were dramatically reduced in the dentate gyrus to only ∼10% of controls’ levels, whereas MAP 2c, which labels immature neurons, showed normal abundance. 36 In layer 2 of the EC, there were 64% and 58% fewer NeuN+ (mature) neurons in MCI and AD, respectively, and a corresponding 24% and 25% loss of volume. 37 In patients with MCI, smaller volumes at baseline of the hippocampus and EC, reflecting more neuronal loss, were associated with the risk of conversion to AD over 5 years. 38 In AD, neurons in the EC are apoptotic. 39 Similar to AD, mice with knockouts for PS1 and PS2 developed neurodegeneration and concomitant neural regeneration in the dentate gyrus, but, importantly, there was impaired survival of the newly generated neurons. 40 Also in transgenic mice, the newly differentiating neurons expressing BrdU and DCX in both the SVZ and GCL were reduced ∼2-fold. 32 Likewise, by age 3 months and about 1 year before either amyloid deposits or neurofibrillary tangles were seen, transgenic mice carrying AD-related genes showed degenerating neurons and axonal swellings that contained abnormal amounts of microtubule-associated proteins. 41 The brains from patients with AD showed axonal swellings like those seen in the mice. 41,42
Decreased migration and survival of mature neurons
In MCI, smaller volumes of the hippocampus and EC at baseline, presumably reflecting either less neuronal migration or migration but with subsequent neuronal death, were associated with the risk of conversion to AD over 5 years of follow-up. 38 Young mice transgenic for PS 1 and 2, which cause neurodegeneration, showed enhanced neurogenesis in the SGZ, but in older mice, there was migration of mature neurons to the hilus and granular cell layer where BrdU labeling demonstrated decreased survival. 40 This study demonstrated 2 points: first, that neurodegeneration triggers enhanced neurogenesis and second, neural survival is ultimately decreased. The latter was also shown in the superior temporal sulcus, where neurons were 53% fewer in AD than controls. 35
A reasonable interpretation of the above data is that proliferating neurons in brains of both MCI and AD die in great numbers either before or after their maturity, and this is more as the dementia progresses.
Blocks in the Genesis of Astrocytes in AD Are Beyond Those in Normal Aging
Astrocytes account for about 50% of cerebral volume. They direct stem cells toward neurogenesis 3 and may act as NSCs. 4 Astrocyte numbers in the dentate gyrus were more reduced in Braak stages 3 to 4 than in 0 to 2. 43 In mice transgenic for APP, PS, and tau, astrocytes in the EC showed generalized atrophy, with major reductions in their primary, secondary, and distal branches. These changes appeared in 1-month-old animals before there were any signs of AD pathology, indicating that astrocytic atrophy is a very early pathogenetic event. 44 And in AD itself, there were more senescent astrocytes in the frontal cortices than in controls. 45 Thus, in AD, there is a reduction in astrocytes in the early stage of the disease.
Changes in Capillaries in AD Are Beyond Those in Normal Aging
In AD, the basement membrane of cerebral capillaries showed increased width and degenerating pericytes. 46,47 Capillary dysfunction, affecting cerebral blood flow (CBF), permeability of the BBB, and thus metabolism of neurons and astrocytes, is an early feature of AD, which had changes in basement membranes of arterioles that were similar in pre-Braak as well as in all later Braak stages. 48 Astrocytes’ processes plaster the capillary network, participating in both the formation of the BBB and the control of CBF. Thus, 76 patients with AD had changes in basement membranes of arterioles that were similar in pre-Braak as well as in all later Braak stages. 48 In AD, the percentage volume of capillary endothelial cells that is occupied by mitochondria is substantially reduced, 49,50 which would substantially reduce the available energy required for adjusting CBF. Other studies saw reduced resting, CBF, and a delayed response of CBF in both MCI and AD. 51,52 In the Rotterdam Study, more subsequent cognitive decline occurred in those having low CBF velocity at baseline. 53 In all the affected areas of the brain in AD, string vessels, which are remnants of capillary injury, are increased, 54 vascular density is reduced, 55 and compensatory new vessel formation occurs. 56 Although the number of capillaries in the SGZ was markedly reduced with age, 21 scanning electron microscopy of the whole brains of young mice transgenic for AD showed abnormal microvessels before the histopathologic appearance of the disease, indicating an early role in pathogenesis. 57 Repair of damaged capillaries requires vasculogenesis, which involves endothelial progenitor cells that are fewer in AD, 58,59 implying vasculogenesis insufficient to cover capillary loss. In AD, the deep white matter showed capillary strings, and in the cerebral cortex, an age-related decline in capillary density. 54
Part 2: The Subcellular Pathogenetic Mechanisms
Figure 1 illustrates the subcellular processes and their interactions, most of the evidence for which was obtained in studies of neurons but it is likely that it applies also to astrocytes; some data applying to cerebral capillaries will be mentioned below.
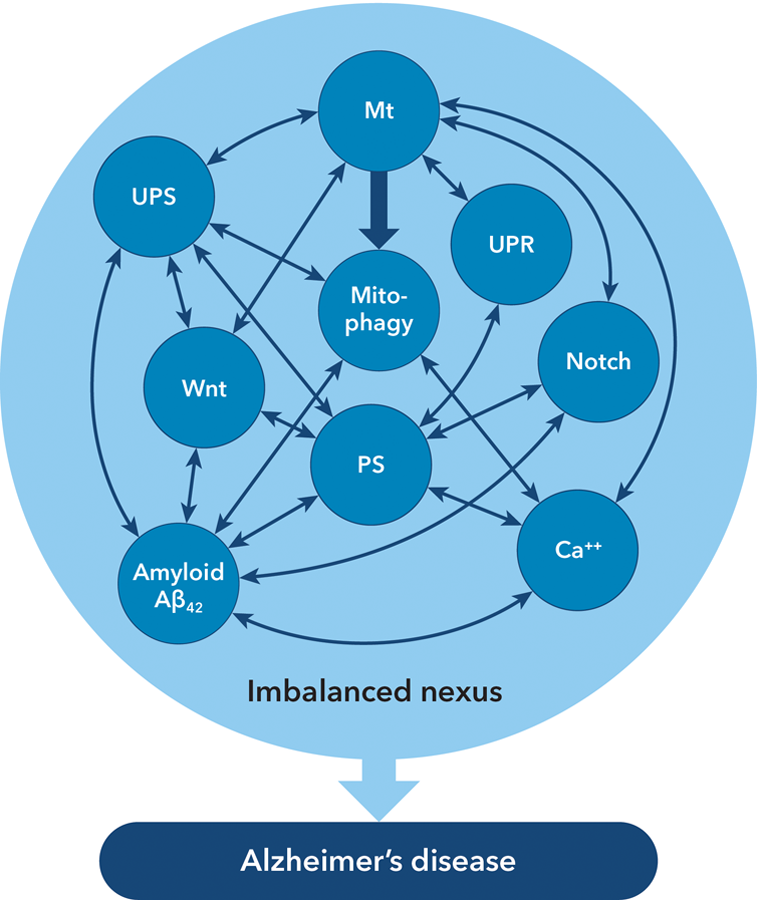
Multiple interactions between the subcellular mechanisms affecting neurons, astrocytes and capillaries in Alzheimer’s disease.
The UPR
Proteins must be folded correctly for their normal functioning; accumulation of unfolded or misfolded proteins induces the UPR as an adaptive response. Incorrect folding of proteins may produce amyloid, so the UPR is important for AD and is activated early. 60 The UPR increases transcription of genes encoding the so-called chaperones, for example, GRP78, which facilitate protein-folding. GRP78 reduced formation of the polypeptide Aβ42, which is a precursor of amyloid fibrils and plaques; thus, the low levels of GRP78 found in AD brain would enhance generation of Aβ42 and amyloid plaques. 61 In brief, the UPR is decreased in AD, leading to accumulation of unfolded or misfolded proteins including amyloid.
Presenilins
The amyloid precursor protein (APP) is processed by the enzymes α-, β-, and γ-secretases. The PS1 is a component of γ-secretase. 62 α- and β-secretases generate the carboxy-terminal fragments of APP, from which PS1 generates Aβ peptides. 63 The FAD is caused by mutations in several genes, especially the PS1 gene (small). 63 At first glance, it seems paradoxical that mutant PS1 (PSImut) in FAD would have enhanced rather than decreased formation of amyloid. The explanation seems to be that its deficient cleavage activity causes more of the amyloidogenic longer polypeptide Aβ42 and reduction of the shorter, less amyloidogenic Aβ39 and Aβ40 because formation of the latter requires further cleavage of Aβ42. 64 The PS1 also cleaves Notch, so PS1mut would impair the beneficial effects of Notch signaling (see below) on neurogenesis. 65
Amyloid
Evidence for amyloid’s involvement in the pathogenesis of AD is very strong, and it is widely held that either amyloid or its precursors, the Aβ peptides or oligomers formed from them, are the major cause for AD. Certainly, amyloid or its oligomers may usually be the necessary causes, but the data show that their presence may not always be sufficient to explain AD. Reduced number of neurons in the EC of AD, the main site of neuropathology in AD, was related to neither number 34 nor volume 66 of amyloid plaques. Over 35% of cognitively normal elders have those plaques in their brain 67 ; further, a study showed that cognitive reserve weakens the relationship between amyloid deposition and cognition. 68 If it is oligomers not plaques that cause the dementia, then it is relevant that oligomeric Aβ was present in 99 of 100 of the dense cores of amyloid plaques in mice transgenic for mutant APP, 69 because, in a clinical trial, when the plaques were eliminated by antiamyloid antibodies, those oligomers would have been released and cognition should have worsened but did not do so. 70 Another concern is that oligomers stick to other proteins, 71 so that synaptic and other toxicity, shown in many reports to be induced by their direct application to brain slices or cultured neurons, might be nonspecific. Finally, intracellular reaction rates are affected by the molecular crowding that occurs when large molecules occupy space so that other molecules become more concentrated. Concentration and therefore their biological activities of cellular and extracellular reactions can increase by 2 orders of magnitude as the molecular weight of an introduced agent exceeds 50 kDa 72 and the molecular weight of Aβ oligomers can be >100 kDa. 73 For that reason, the effects on animal behavior after changing oligomers’ concentration, or of oligomers in human AD, might be due not only to a direct effect but also to an indirect effect on neurotransmission caused by molecular crowding. In brief, amyloid and its oligomers surely have important roles in causing AD but most likely act as participants with other processes in a multicausal nexus.
Mitochondria
There are many hundreds of mitochondria in every cell and they create most of the energy required for cellular, including neuronal, function. Besides, mitochondrial damage may induce formation of harmful, reactive oxygen species. Brain mitochondria in AD were reduced in number, 74 were morphologically abnormal, 75,76 had fewer genes encoding subunits of the electron transport chain, 77 had decreased activities of enzymes in the TCA cycle, 78 and at-risk ε4 carriers had lower cytochrome oxidase activity in the posterior cingulate cortex. 79
Mitochondrial morphology is constantly changing by the processes of fusion with neighboring mitochondria and fission into daughter mitochondria. Fusion of 2 mitochondria creates beneficial interaction between the proteins of the 2 organelles. In AD brains, fusion proteins were substantially reduced and fission protein levels were almost 5-fold higher. 80 Fission segregates segments of damaged mitochondria for their degradation in the UPS (see below), but excessive fission releases caspase-activating proteins that cause apoptosis of neurons, astrocytes, and endothelial cells. 81
The UPS
The UPS is a mechanism, besides autophagy (see below), for disposal of dysfunctional cellular proteins that may cause neuronal death. Ubiquitin is a small peptide that becomes conjugated by a ligase to the protein requiring disposal; the ubiquitin–protein complex enters a proteasome vacuole, to be degraded by proteolytic enzymes. In AD brain, there is defective ubiquitination 82 and a 48% decrease of proteasome activity in the hippocampus. 83 Mutant ubiquitin, seen in 100% of patients with early and advanced AD, 84 caused neuritic beads that contained mitochondria. 85
Autophagy, Mitophagy, and Mitochondrial Biogenesis
Autophagy is also a mechanism that delivers unwanted cellular materials to lysosomes for degradation. In mitophagy, damaged mitochondria enter an autophagocytic vacuole (AV) that fuses with a lysosome and is digested. Mice whose neural cells had defective autophagy developed progressive behavioral deficits. 86 In AD brains, there were 20-fold more AVs in the neuritic processes than in controls. 87 Beclin 1, a mediator of autophagy, was decreased in AD brains early in the disease, reflecting inadequate autophagy/mitophagy. 88 Mitophagy is important because mitochondrial depletion stimulates biogenesis of new, healthy organelles.
Mitochondria can replicate because their origin is bacterial, and therefore, they contain their own genome. The peroxisome proliferator-activated receptor-γ coactivator-1α , a major regulator of mitochondrial biogenesis, induces genes for transcription and replication of mtDNA. By inducing mitochondrial biogenesis, it protects neural cells from oxidative stressor–mediated death. 89 Those beneficial effects are absent in AD, where its level is decreased. 90
Wnt/β-Catenin Signaling
Wnt/β-catenin signaling affects most metabolic functions of cells. Among its several signaling pathways, one leads to gene transcription and another affects intracellular calcium. After its release from the complex with Wnt, β-catenin then translocates to the nucleus, where it activates genes that transcribe proteins participating both in the formation of dendrites and synapses and also in axon guidance. β-Catenin is destabilized by PSImut in FAD, and β-catenin levels are decreased in brains of patients with AD with PSImut. 91
Notch
Notch proteins are important in determining neuronal cell structure and play a critical role in maintaining NSCs 92 and controlling neurogenesis. 93 Expression of Notch was increased by 21-fold in hippocampal sections of AD brains, 94 which is presumably a compensatory reaction to the decreased number of neurons. In brief, Notch maintains NSCs and regulates neurogenesis.
Tau
Tau is a microtubule assembly factor that is expressed abundantly in the central and peripheral nervous systems. Hyperphosphorylated tau interacts poorly with microtubules, which is disadvantageous because hyperphosphorylated tau cannot promote proper microtubule assembly. Hyperphosphorylation occurs in response to various stressors, tends to aggregate and cause neurotoxicity, and is a major component of the neurofibrillary tangles in AD. In brains from advanced AD (Braak groups 1V-V1), the amount of phosphorylated tau was increased. 41 Microtubules act as rails along which mitochondria and proteins are transported down the axon; an indication of microtubules’ importance is their reduction by 6-fold in number and 4-fold in length in pyramidal neurons from patients with AD. 95 On the other hand, whether or not tau dysfunction is an early or later factor in pathogenesis is still debatable. One theory holds that Aβ is upstream of tau pathology. However, studies in tau knockout mice show, overall, that tau reduction prevents Aβ-mediated deficits. 96 Although the mouse brain is significantly different from that of humans, a study of both transgenic mice carrying AD-related genes, and of AD itself, showed that by 3 months of age and about 1 year before either amyloid deposits or neurofibrillary tangles were seen, neurons were degenerating and neurons of both the mice and of AD itself had axonal swellings containing abnormal amounts of microtubule-associated proteins. 41 Because of these uncertainties about the timing of its role, tau is not included in Figure 1. Another paradox is that among those phosphatases that regulate phosphorylation of tau, the major one is protein phosphatase-2A and this is downregulated in AD brain. 97
Calcium
Calcium disposition in the brain and its changes in both normal aging and AD is highly complex and reviewed elsewhere. 98 A hypothesis proposed 27 years ago suggested that sustained disturbance of calcium homeostasis is a proximal cause of neurodegeneration in AD. 99 Every major gene that causes increased susceptibility to AD alters intracellular calcium signaling. 98 In particular relation to AD, the PS induce calcium release from the endoplasmic reticulum (ER) and this enhances Aβ generation. 100,101 And ER stress that leads to release of calcium activates various kinases and proteases involved in autophagy/mitophagy signaling of dendrites and synapses and in axon guidance. 102 In brief, abnormal calcium signaling plays a major role in AD.
Part 3: Interactions Between the Subcellular Mechanisms of Pathogenesis
Subcellular Interactions in Neurons in AD
Although many interactions are between 2 of the subcellular mechanisms, there are numerous interactions between 3, 4, or even 5 of them. Figure 1 illustrates the complexity of the interactions. The more the number of processes involved in a mutual interaction, the greater the likelihood that dysfunction of 1 process might impair the entire group.
Amyloid interactions
With mitochondria: Amyloid deposits in mitochondria impair mitochondrial and neuronal functions. The Aβ-induced autophagy in neurites and mitochondria were present in the autophagosomes. 103 The importance for AD is that impaired mitochondrial functions have deleterious effects upon every aspect of brain function.
With UPR: Impairment of the UPR prevents proper folding of proteins, so enhances amyloid formation.
With Wnt signaling: The neurotoxicity of Aβ was reversed by raising β-catenin levels 104 and by the Wnt-3a ligand, 105 which beneficially induced mitochondrial biogenesis. 106 The implications of this are counteractive to AD.
With calcium: Neurites adjacent to amyloid plaques had calcium overload, which would produce disruption of neuronal networks. 107 Consequences for AD are obviously serious.
Presenilin interactions
The PS have many interactions with components of the causal nexus including APP generation, Wnt/β-catenin signaling generation, Notch signaling generation, and UPR generation 108 (also see Table 1 in the study by Verdile et al 62 ). There are multiple ramifications of these several interactions, some antagonistic to and others furthering the occurrence of AD.
PSImut produces many impairments: PSImut-expressing neurons had decreased induction of GRP78 (which facilitates protein folding in the UPR), therefore less protein folding, and brains of AD had decreased GRP78.
61
The implication of this is that amyloid formation may be enhanced. PS1mut-expressing cells had 70% less of the enzyme, pIRE1, an upstream component of the UPR.
61
This also might lead to enhanced formation of amyloid. Although PS1wt can “leak” calcium and favorably affect neuronal function, PS1mut cannot do so.
101
PS1mut inhibited Notch processing as compared with PS1wt.
109
Notch processing is critically important for neuronal biogenesis.
Mitochondrial interactions
With Aβ: Dysfunctional mitochondria reduces formation of ATP, which impairs correct protein folding and contributes to the formation of amyloid. 110 Reduced formation of ATP affects negatively almost every biological reaction in the brain so inevitably is deleterious for AD by the negative effects upon neuronal, astrocytic, and capillary functions.
With mutant Aβ: In neuronal cells, mutant Aβ produced increased mitochondrial fission, loss of dendritic spines, and cell death. 111 All of these effects would worsen AD.
With Wnt: Wnt induces mitochondrial biogenesis. 106 This effect is beneficial for AD.
With ER and calcium: The ER is the main cellular site for calcium storage. Mitochondrial-associated membranes are contacts between mitochondria and ER; they are upregulated in AD brains 112 and calcium signaling results. 113,114 Intracellular calcium release, whether within cytosol or intramitochondrial, has multiple repercussions for neuronal function that might affect AD adversely.
With UPR: Separate UPR formats exist in different subcellular compartments, with 1 specifically in mitochondria (UPRmt), which is important if mitochondrial dysfunction causes accumulation of unfolded proteins. 115 This is another way by which amyloid formation may be enhanced and affect AD negatively.
With UPS: Several UPS components regulate mitochondrial dynamics, degradation of dysfunctional mitochondria, and mitophagy (see Table 1 in the study by Franz et al 116 ), with a cross talk between UPS and mitophagy because both systems use ubiquitilation. 115,116 Ramifications of impaired UPS are largely harmful for AD.
With Notch: See below.
The UPR interactions
With mitophagy: As noted, there is cross talk between UPS and mitophagy, linked by their common use of ubiquitilation. 115,116
With tau: Activated UPR increases tau phosphorylation. 117 Hyperphosphorylated tau detracts from microtubule formation, leading to neuronal dysfunction and therefore affects AD negatively.
With autophagy: Cross talk occurs between UPR and autophagy. 118
With ubiquilin variant: The ubiquilin molecule contains some sequences similar to ubiquitin. Ubiquilin binds PS 119 and is abundant in brains of mice transgenic for APP and PSmut. 120 A ubiquilin variant confers increased risk for AD 121 and WT ubiquilin levels are decreased in AD brains. 122 The implication for AD is impaired UPR and UPS, with heightened liability to amyloid formation.
With Aβ and tau: UPR interactions with Aβ, and with tau, inhibit proteasome function, 123 -125 with accumulation of Aβ, Aβ oligomers, and tau. 125 Each of those accumulations is harmful for AD.
With autophagy: UPS and autophagy act reciprocally; downregulation of one is accompanied by upregulation of the other. 126 Balanced reciprocal effects are neutral for AD, but imbalanced reciprocity would be detrimental for AD.
Calcium interactions
With mitophagy: Either high or low calcium stimulates mitophagy. Since mitophagy leads to mitochondrial biogenesis, this is counteractive to the progression of AD.
With chaperones: Calcium acts as a protein-folding buffer in the ER and affects chaperone function. Improved protein folding is also beneficial for AD.
With mitochondrial biogenesis: Calcium-dependent signaling affects mitochondrial biogenesis. 127
With PSImut : Both PSImut and PS2wt enhance release of calcium from ER, impairing neuronal function, 101,128 that is, this could exacerbate AD.
Wnt interactions
With Wnt ligands: Wnt ligands cause β-catenin to translocate into the nucleus and activate genes. 129 Overall, the products of those genes counteract the progression of AD.
With PS1 and Aβ: PS1 cleaves APP to yield Aβ that stimulates degradation of β-catenin. Each effect would enhance the progression of AD.
With tau: Inhibition of canonical Wnt signaling by DKK1, elicited tau hyperphosphorylation, and cell death in cultured neurons. 130 These effects are all permissive of AD.
Notch interactions
With PS: PS1 is required to cleave and activate Notch.
With Wnt: Wnt reduced phosphorylation of Notch caused by GSK-3β. 131 Phosphorylation impairs the beneficial efficacy of Notch signaling, so this is another way by which Wnt signaling serves to neutralize AD.
With mitochondria: Cells exposed to the pro-apoptosis molecule, Bax, had prolonged survival involving a Notch-derived protein that depended upon the presence of certain mitochondrial proteins. 132
With both Aβ and epidermal growth factor (EGF): Interplay between Notch and EGF in the SVZ of the third ventricle affects both NSCs and NPC, 133 and EGF enhanced the neurotoxicity of Aβ. 134 Effects upon AD depend upon the balance of the interaction.
Tau interactions
With chaperone-mediated autophagy: The chaperones HSP70 and HSP90 reduced tau phosphorylation, 135 so that upregulation of chaperones may suppress formation of neurofibrillary tangles caused by hyperphosphorylation of tau. HSP70 binds tau and inserts it directly into the lysosome for degradation. 136 Reduction of tau phosphorylation would decrease both liability to and progression of AD.
With UPS: The defective ubiquitination seen in AD brain 82 might be caused in part by accumulation of tau because tau aggregates, that occur due to hyperphosphorylation, impair proteasome activity. 137
Interactions in neurons between 3, 4, and 5 elements
Mitochondria, UPS, and microtubules: Miro, a component of a complex affecting mitochondrial movement, influences mitochondrial transport along neuronal microtubules. 116 Phosphorylation triggers Miro’s degradation by the UPS, thus disturbing mitochondrial transport and function. 138 PS, Aβ generation, Wnt/ β-catenin, Notch signaling, and UPR all mutually interact. 62
Between Aβ, UPS, dysfunctional mitochondria, and autophagy: ROS from dysfunctional mitochondria produces 4-hydroxynonenol (4-HNE), a product of lipid peroxidation. Both WT and mutant Aβ caused 2-fold increases in HNE, which decreased autophagic enzymes (trypsin, chymotrypsin, cathepsins), but mutant Aβ also caused increased chaperone activity of HSP90, suggesting adaptive compensation to the decreased autophagy. 126
Subcellular Interactions in Astrocytes in AD
Astrocytes and UPR: Some but not all antidepressants stimulate autophagy in astrocytes. 139
Astrocytes and UPS: The UPS is more active in astrocytes than in neurons, 140 implying a more important role for astrocytes in maintaining protein formation.
Astrocytes and amyloid: Cultured astrocytes have raised calcium levels after exposure to Aβ, and network-wide, interastrocytic elevations in resting calcium start adjacent to local amyloid plaques and are thought to be due to propagation of intercellular calcium waves. 107
Astrocytes, amyloid, and mitochondria: Astrocytes from the posterior cingulate cortex in AD are associated with downregulation of the mitochondrial gene FASTKD2 that is involved in regulating apoptosis and with upregulation of TRMT61B that methylates adenosine at position 68 of mitochondrial transport RNAs, which are required for respiratory factors. 141 Also upregulated are immune response genes, including clusterin, that encode a chaperone which inhibits Aβ uptake by astrocytes so they may affect Aβ clearance. 141
Astrocytes and autophagy/mitophagy: Interestingly, mitochondria in axons of retinal ganglion cells were shown to be taken up by astrocytes in which mitophagy then occurred. 142 It is unknown whether transcellular mitophagy occurs in other circumstances.
Astrocytes and calcium: Intracellular calcium is critical for intracellular communication in the tripartite synapse. In cultured astrocytes, the NMDAR mediated release of ionized calcium from intracellular pools. 143
Interactions between 3, 4, and 5 elements
Interactions between astrocytes, calcium, PS1, and amyloid: Human neuroglioma cell lines, transfected to overexpress APP and treated with the calcium channel blocker (CCB) nifedipine, had a 40% reduction in levels of PS1 and decreased levels of Aβ1 to Aβ42, suggesting possible mechanisms linking astrocytes with AD—although the evidence that CCB drugs reduce decline in cognition is controversial. 144 Raised calcium levels occur in cultured astrocytes after exposure to Aβ, network-wide, interastrocytic, elevations in resting calcium start adjacent to local amyloid plaques and are thought to be due to propagation of intercellular calcium waves. 107
Subcellular Interactions in Endothelial Cells in AD
Interaction with UPR: Incubation of endothelial cells with Aβ1-40 increased the levels of several markers of the UPR. 145
Interaction with mitochondria: Although not performed in AD, studies show the importance of mitochondrial functions for the proliferative capacity of vascular smooth muscle, which is a major element controlling CBF.
Part 4: Potential Treatments
Drugs suggested as likely to impede the progression of AD are those that are both currently available and significantly affect the elements described above as acting early in pathogenesis. Potential treatments include lithium, valproate, antidiabetic drugs, prazosin, and erythropoietin, besides the currently approved acetylcholinesterase inhibitors. The foregoing drugs are the ones for whose efficacy the evidence is reasonably abundant. There are others that warrant only passing mention, but for those the evidence for efficacy is either not robust or they have serious toxicities, for example, methylene blue, taxol, and phosphodiesterase inhibitors.
Lithium, Valproate, and Fluoxetine
Data suggest that lithium and valproate might be clinically effective in treating AD by interrupting the blocks to neuronal renewal. A series of publications 146 -149 demonstrated that both lithium and valproate affect several mechanisms. Lithium, by inhibiting GSK-3, reduces tau phosphorylation that is mediated by GSK-3. Moreover, lithium rescues β-catenin levels, thus Aβ-induced hippocampal neurodegeneration 105 and lithium upregulates Bcl-2, which is cytoprotective, antiapoptotic, and neuroprotective. 150 Valproate inhibits GSK-3 and is also a histone deacetylase inhibitor. Evidence suggests that lithium and valproate have synergistic neuroprotective effects. 147 Another beneficial effect of valproate is elongation of the processes of cultured astrocytes, which would counter the atrophy seen in astrocytes of AD. 147 Valproate induces clusterin (apolipoprotein J), which blocks transport of Aβ across the BBB, and by decreasing the brain’s concentration of Aβ, it prevents Aβ aggregation and fibrillization. 151 Also benefitting capillaries, valproate causes increased production of vascular endothelial growth factor, facilitating endothelial cell proliferation and increasing CBF. 152 Since astrocytes have a central role in neuronal functioning, it is worth mentioning that various antidepressant molecules, for example fluoxetine, improve the morphology of astrocytes. Treatment with fluoxetine enhanced the number of astrocytes’ processes 11 and a small study showed cognitive improvements in patients with MCI receiving fluoxetine as compared with controls receiving placebo. 13 Valproate stimulates release of various neurotrophic factors, for example BDNF from astrocytes. 146 -148
Prazosin
The evidence is moderately convincing that prazosin may be an effective treatment and it benefits all 3 of the cells and structures affected early in pathogenesis.
The locus coeruleus is the major noradrenergic nucleus in the brain. It contains at most 50 000 noradrenergic neurons and the number is reduced early in AD. 153 Its adrenergic neurons project to the EC, hippocampus, and thalamus. In the brains of 46 persons with dementia as compared with 33 controls, α-2 adrenergic receptor density was 50% less in cells of the rostral locus coeruleus, and the reduction in the noradrenaline concentration in the cerebral cortex correlated with cognitive impairment. 154 Experimental lesions of the locus coeruleus exacerbated AD-like neuropathology and cognitive deficits in several transgenic mouse models of AD. 155 In live mice, both direct stimulation of the locus coeruleus and injection of an α-adrenergic agonist created rapid increases of calcium in astrocytes. 156 Noradrenergic mechanisms appear to play important roles in modulating the activity of the basal cortical cholinergic system and its response to injury and in modifying cognitive functions including memory and attention. 157 Mechanisms by which noradrenaline may protect or promote recovery from neural damage include affecting neuroplasticity, neurotrophic factors, neurogenesis, inflammation, cellular energy metabolism and excitotoxicity, and oxidative stress. In addition to neurogenesis, an α-2 adrenergic receptor agonist caused gliagenesis 158 and also improved synaptic plasticity, cognition, and mood in mice transgenic for an α-1 adrenergic receptor. 157
Several trials were made in patients with AD using clonidine, an α-2 adrenergic receptor agonist, but results were either weak or negative. 159 Prazosin, an α-1 adrenoreceptor antagonist, was given in a study involving 22 patients with probable or possible AD, who were randomized 1:1 to either prazosin or placebo. Over an 8-week period, there were significant improvements in the Brief Psychiatric Rating Scale, Neuropsychiatric Inventory, and the Clinical Global Impression of Change. 160 Prazosin was also given to transgenic mice carrying the APP Swedish double mutant and gave beneficial results, namely, an increase in numbers of astrocytes, increase in hippocampus neurons, a decrease in levels of Aβ, and prevention of memory deficits. 161
The difference between prazosin and clonidine in their effects is worth exploring; however, adrenergic mechanisms in the brain are extremely complex because effects differ not only according to the brain area but also because there are several subtypes of α receptor, each of which responds differently to its ligation 159,162 ; in addition, memory itself is not a single function. Thus, the following brief explanation is, necessarily, a simplification.
Activation of α-1 adrenoreceptors may impair the function of the prefrontal cortex, and infusion of an α-1 adrenoreceptor agonist impaired working memory in animals. 162 So prazosin, an α-1 adrenoreceptor antagonist, would be beneficial. Clonidine, on the other hand, inhibits the firing of norepinephrine cell bodies in the locus coeruleus, 159,162,163 which is deleterious. However, the complexity is illustrated by the finding that clonidine impaired short-term recognition 164 but enhanced working memory in AD. 165 Another adrenergic influence is upon gap junctions, which are intercellular channels that allow the passage of ions and small molecules between neighboring cells. This provides a link between neurons, astrocytes, and blood vessels. 166,167 Gap junctions can be up- or downregulated by signal transduction pathways, one of which is initiated by the α-1 adrenoreceptor that, when activated, closes the gap junction. Closure, which is initiated by noradrenaline and prevented by prazosin, an α-1 adrenergic receptor inhibitor, 166 would impair communication between neurons, astrocytes, and blood vessels. Clonidine, an α-2 adrenergic agonist, inhibits the actions of noradrenergic neurons projecting from the locus coeruleus to the cerebral cortex. 163 In brief, according to the above difference in their effects, one would expect prazosin to be beneficial and clonidine to be deleterious, for brain function.
Insulin and Antidiabetic Drugs
Insulin and thiazolidinediones benefit neurons; the evidence is moderately convincing that such drugs may be an effective treatment. After it had been noted that there are many insulin receptors in the brain, it was suggested that insulin might have a role in cognition. A study that measured insulin levels in patients with AD found that those who were not apoE4 homozygous had high plasma levels of insulin, and this was more pronounced in those with advanced disease. 168 Raising plasma levels by either intravenous or transnasal application improve cognition in both cognitively normal patients and those with AD. 169,170 Soluble Aβ oligomers were shown to downregulate insulin receptors; and the oligomers were prevented from binding to synapses in the hippocampal nerve cell culture, by either insulin itself or rosiglitazone, which is an insulin-sensitizing drug. 171 A study comparing cognition in well-matched patients with either AD alone or both AD and diabetes, 85% of whom were using antidiabetic drugs, found less impairment of cognition in those having both conditions. 172 Inhibition of GSK-3β has an insulin-sensitizing effect 173 ; it is worth noting that thiazolidinediones, for example rosiglitazone, are inhibitors of GSK-3β. 174 Other studies, including 1 of 511 patients, confirmed the beneficial effects of rosiglitazone on cognition, 175,176 although a prospective trial in 693 patients did not show benefit. 177 Rosiglitazone significantly prevented cognitive impairment induced by status epilepticus. 178 In mice transgenic for AD, rosiglitazone attenuated learning and memory deficits. 179
Pioglitazone is another insulin sensitizer. In a prospective cohort study of 145 928 patients aged ≥60 years, who at baseline were free of dementia, long-term use of pioglitazone reduced the dementia risk by 47%. 180 And in a 6 months’ trial of 21 patients with both mild AD and diabetes, versus 21 controls, the group given pioglitazone showed improved cognition. 181 In a mouse model of AD, pioglitazone improved memory and also decreased astrocytic activation. 182
Erythropoietin
The evidence is substantial that erythropoietin may be an effective treatment; it benefits all 3 of the cells and structures affected early in pathogenesis. In humans, erythropoietin crosses the BBB, 183 and receptors for erythropoietin are present on neurons, astrocytes, and both endothelial cells and pericytes, that is, all of those cellular elements that are impaired early in the pathogenesis of AD. Beneficial effects of erythropoietin in the brain include those on memory and behavior, both cholinergic and adrenergic neurons, astrocytes, capillaries, and the BBB. And it protected neurons from degeneration after injury, 184 presumably because it has a neurotrophic function. It promoted the regeneration of both cholinergic 185 and adrenergic neurons. 186 In vitro, neuronal differentiation was induced by coculturing NSCs with astrocytes; further differentiation occurred when erythropoietin was added to the medium. 187 In rats with embolic stroke, treatment with erythropoietin significantly improved the neurological outcome. 188 Those animal studies showing neuroprotection were confirmed in humans by a double-blind study of erythropoietin administered to patients with strokes. 189
In humans, treatment with erythropoietin improved behavior in patients with schizophrenia and multiple sclerosis 190,191 ; and when administered before and after coronary artery bypass grafting, there was a trend (P = .085) toward neurocognitive improvement. 192 Patients were shown pictures after receiving either erythropoietin 50 000 U or saline; 1 week later during functional MRI when they were again shown those pictures, patients given erythropoietin had substantially improved visual recognition. The left and right hippocampus were activated in all patients. Independent of any change in hematocrit, erythropoietin significantly enhanced the hippocampal response, and analyses of whole brain showed that the drug influenced a broad neuronal network engaged in encoding and recognition of visual scenes. 193 The same group reported a similar study in patients with depression using the same dose of erythropoietin. Those given erythropoietin showed improved memory compared with those given saline and, again, there were no changes in hematocrit. 194
In a mouse model, erythropoietin induced improvements in several aspects of memory including long-term potentiation, a cellular correlate of learning processes in the CA1 region of the hippocampus; the improvement was unrelated to changes in hematocrit and persisted for 3 weeks after termination of EPO injections. 195 A single dose in young rats provided prolonged behavioral benefit, 196 and a favorable effect of the drug was also seen in astrocytes from both mice and rats. 197 -199 The drug mobilized endothelial precursor cells from the bone marrow 200 ; in rats with embolic stroke, the density of microvessels at the stroke’s boundary was increased 188 ; in vitro studies showed enhanced capillary tube formation from cerebral endothelial cells 188 ; in the hippocampus, antierythropoietin staining was primarily localized to capillaries 183 ; and in rats, a leaky BBB caused by ischemia–reperfusion was decreased by erythropoietin. 201
Conclusions and Summary
The AD has a multicausal pathogenesis: at the macrocellular level, there is intimate anatomic interaction between neurons, astrocytes, and capillaries and a failure to renew those cells; at the subcellular level, many of the causal elements interact with each other. Because of the several interactions between the subcellular mechanisms (Figure 1), the use of combinations of agents will more likely yield success than the use of single agents. Available drugs that should be considered in a clinical trial include lithium, valproate, pioglitazone, erythropoietin, and prazosin. The success that has to date eluded the use of antiamyloid antibodies in established AD might be achieved if the antibodies were administered with 1 or more of the above drugs.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.