
Abstract
Aim:
The aim of this study is to extend the molecular mechanism of Tong Luo Jiu Nao (TLJN) for Alzheimer’s disease (AD), which is a modern Chinese formula that has been used to treat AD.
Methods:
The senescence-accelerated mouse prone 8 strain (SAMP8) is one of the most appropriate models to study the mechanism that underlies AD. The levels of plasma amyloid β (Aβ) and the Aβ deposits were measured using enzyme-linked immunosorbent assay and immunohistochemistry. Immunoblotting was used to observe the effect of TLJN on inflammatory mediator expression in an senescence-accelerated mouse model of AD.
Results:
Our data showed that the TLJN-treated groups exhibited a reduction in plasma Aβ levels and reduced Aβ expression. Moreover, TLJN effectively attenuated Aβ-induced activation of extracellular signal-regulated kinase and c-Jun N-terminal kinases and blocked changes in inflammatory mediator expression.
Conclusion:
These data suggest that TLJN might have protective effects and could potentially act to attenuate inflammatory stress in the pathogenesis of AD.
Introduction
Alzheimer’s disease (AD) is a progressive neuronal degenerative disorder of the central nervous system that culminates in the loss of cognitive function. 1,2 The pathological hallmarks of AD include the accumulation of neurofibrillary tangles, the formation of extracellular amyloid β (Aβ), and inflammation. In tissues that surround Aβ deposits, an inflammatory response is apparent. Many studies have implicated cerebral microvascular Aβ deposition in promoting neuroinflammation in AD. 3 –6 An additional feature in the brain of patients with AD is the presence of activated microglia and astroglia, which release proinflammatory cytokines and chemokines. In the brain of patients with AD, both the presence of inflammatory mediators and the increased expression of the full complement cascade in the vicinity of Aβ deposits strongly indicate the contribution of inflammation to AD pathogenesis. 7,8 Many adhesion molecules and chemokines are involved in the neuroinflammatory cascade. This complex network of pathways is mostly conditioned by adhesion and adhesive agents, cell chemotaxis, and chemotactic mediators. Herein, the expression levels of inflammatory mediators in a mouse model of AD were assessed.
Traditional Chinese Medicine has been widely used for the clinical treatment of neuronal degenerative disease. One traditional Chinese medicinal substance, Tong Luo Jiu Nao (TLJN), is composed of several compounds, including geniposide, geniposidic acid, and ginsenoside Rg1. Previously, TLJN was successfully tested in a phase I clinical trial (Chinese SFDA: 2004L01620) designed to test the compound for effects on vascular dementia. 9,10 Currently, a multicenter, double-blinded, randomized, and controlled phase II clinical trial is ongoing. Furthermore, it has been reported that TLJN can improve learning and memory, promote the degradation of Aβ, and clear amyloid plaques from the brain of AD rats. 11 Besides, the compounds in TLJN capable of a reliable curative effect on cerebropathy are actively being pursued. In previous study, it has been shown that geniposide and geniposidic acid had significant neuroprotective effect by improving the expression of neuron trophic factor. 12 It has also been reported that oral administration of ginsenoside Rg1 results in a significant reduction in Aβ levels in the brains of Tg2576 mice, a mouse model of Aβ accumulation. 13 In addition, Ginsenoside Rg1 could also improve the neurological function deficits in adult rat following local ischemia in terms of improving the learning and memory abilities, as well as reducing the apoptosis of neurons in hippocampus CA1 zone. 14
This study aimed to examine the potential neuronal protective effect of TLJN against Aβ-induced neuroinflammation in a senescence-accelerated mouse (SAM) model of AD. The SAM model was established by Takeda and colleagues at Kyoto University by selective breeding from their AKR/J colony in the early 1970s. The SAM subline known as senescence-accelerated mouse prone 8 strain (SAMP8) exhibits an early onset of impaired learning and memory, so it is often used to study aging and age-associated diseases. These mice show natural age-related overproduction of Aβ. Therefore, these animals have been suggested to be a useful model for investigating the events related to Aβ pathology and the early phases of AD. 15,16 We examined the effects of TLJN on reducing Aβ-induced inflammatory stress through reducing the expression of inflammatory mediators. In this study, our data support the possibility that TLJN might have protective effects, including the attenuation of AD pathogenesis and associated inflammation.
Materials and Methods
Animals
Male SAMP8 and senescence-accelerated resistant mouse (SAMR1) mice that weighed ∼25.0 g were purchased from the First Teaching Hospital of Tianjin University of Traditional Chinese Medicine (Tianjin, China). Animals were housed at a density of 6 per cage with free access to food and water. Animals were randomly grouped as control (SAMR1 mice), model (SAMP8 mice), TLJN (SAMP8 mice treated with 9 mL/kg/d TLJN), and positive (donepezil hydrochloride, SAMP8 mice treated with 1.667 mg/kg donepezil hydrochloride). The concentrations used for experiments were based on the daily dose administered to patients in clinical practice. After 21 days, mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) and flush-perfused transcardially with 0.9% saline. Brains were removed and divided at the midline. One hemibrain was postfixed in phosphate-buffered 4% paraformaldehyde (pH 7.4) at 4°C for 26 hours for vibratone sectioning. The hippocampus and cortex were dissected from the rest of the hemibrain, snapfrozen, and stored at –80°C for protein analysis. All animal studies were carried out according to protocols approved by the Institutional Animal Care and Use Committee of Tianjin University of Traditional Chinese Medicine.
Reagents and Chemicals
Anti-Aβ, anti-vascular cell adhesion molecule 1 (VCAM-1), anti-chemokine receptor 5 (CCR5), and isotype control antibodies were purchased from Abcam (Cambridge, United Kingdom). Antiphosphorylated and antitotal extracellular signal-regulated kinase (ERK)1/2 and c-Jun N-terminal kinases (JNK) mitogen-activated protein (MAP) kinase immunoglobulin G (IgG) antibodies were purchased from Cell Signaling Technology (Beverly, Massachusetts). The Aβ1–40, Aβ1–42, and macrophage inflammatory protein 1β (MIP-1β) Enyme-linked immunosorbent assay (ELISA) kits were purchased from Uscn Life Science (Wuhan, China). All other chemicals used were purchased from Sigma-Aldrich (St Louis, Missouri).
The active components of TLJN (Hong Ri Pharmaceutical Company Limited, Tianjin, China) were extracted from Panax notoginseng and Gardenia jasminoides. The TLJN 11,17 was processed following the National Medical Dictionary of China protocol. The main components of TLJN were determined to be geniposide (4.95 mg/mL), geniposidic acid (1.73 mg/mL), and ginsenoside (1.02 mg/mL). The chemical structure of each component and the high-performance liquid chromatography analysis spectrum are shown in Figure 1.
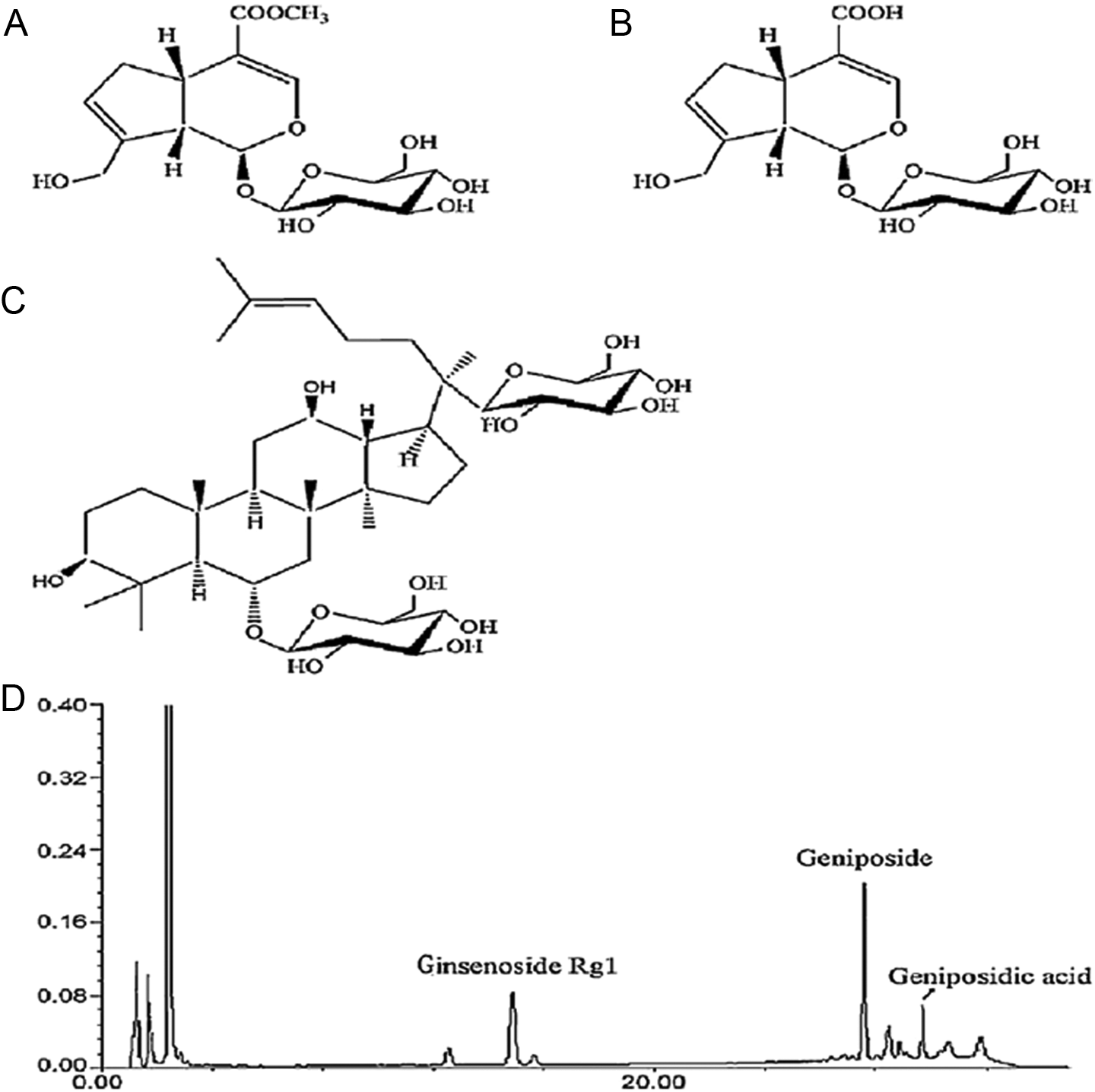
Chemical structures and high-performance liquid chromatography (HPLC) analysis of Tong Luo Jiu Nao (TLJN). A, Geniposide (C17H24O10, molecular weight: 300); (B) geniposidic acid (C16H22O10, molecular weight: 374); (C) ginsenoside Rg1 (C42H72O14, molecular weight: 800); and (D) HPLC analysis of TLJN. The concentrations of geniposide, ginsenoside, and geniposidic acid were equivalent to the corresponding TLJN content, and 7.7 mg/mL TLJN contained 4.95 mg/mL, 1.02 mg/mL, and 1.73 mg/mL of geniposide, ginsenoside, and geniposidic acid, respectively.
Immunoblotting
Brain homogenates from each group of mice were loaded on a 12% polyacrylamide gel and transferred to a nitrocellulose membrane. Immunoblotting analysis was performed as described previously. 18 Brain homogenates (hippocampus and cortex) from each group of mice were used to detect the expression of inflammatory mediators and signal transduction components with the following IgG antibodies: VCAM-1 (1:2000), CCR5 (1:1000), and both antiphosphorylated and antitotal ERK1/2 MAP kinase (1:1000) and JNK MAP kinase IgG (1:1000). As a secondary antibody, goat antirabbit or goat antimouse peroxidase-conjugated IgG antibodies (both 1:5000) were used. In all immunoblotting studies, at least 6 animals were used, and representative data are shown.
Immunohistochemistry
Paraffin-embedded sections were used for immunohistochemistry as described previously. 19 Briefly, sections were deparaffinized in Histoclear (National Diagnostics, Atlanta, Georgia), rehydrated through immersion in a graded ethanol series (100%-70%) to distilled water and washed with 0.01 mol/L phosphate-buffered saline (PBS). Endogenous peroxidase activity was inhibited with 3% hydrogen peroxide in methanol at room temperature for 30 minutes. Sections were incubated with 10% equine serum blocking solution (Hyclone, Logan, Utah) for 30 minutes. Then, sections were incubated with anti-Aβ antibody (1:1000) to identify Aβ deposits. Immunohistochemical analyses were also performed using rabbit anti-VCAM-1 IgG (1:1000) in each group. Staining was visualized using a biotin–avidin peroxide kit (1:50). Sections were counterstained with hematoxylin, coverslipped, and observed under a Leica DM4000 microscope. As negative controls, alternative sections from the same brain were analyzed without incubation with a primary antibody.
Enyme-Linked Immunosorbent Assay
After mice were anesthetized, plasma from each mouse was collected from the endocanthion. The content of Aβ1-40 and Aβ1-42 in plasma was measured using a commercially available ELISA kit according to the manufacturer’s instructions.
Brain homogenates from each group of mice were cleared of cellular debris by centrifugation at 14 000×g. Content of MIP-1β was measured using a commercially available ELISA kit according to the manufacturer’s instructions. Each well was measured using a microplate reader to detect absorbance at 450 nm.
Statistical Analyses
Data were obtained from at least 3 independent experiments and expressed as means ± standard error. Statistical significance was evaluated by 1-way analysis of variance using SPSS version 11.0 (Chicago, Illinois) for repeated measurements. P values less than .05 were considered to be significant.
Results
TLJN Reduces Plasma Aβ Content and Cerebral Aβ Deposition in SAMP8 Mice
Extracellular deposition of Aβ protein in the brain is a prominent pathological feature of AD. Amyloid β peptides are derived by the sequential proteolytic processing of the Aβ precursor protein by β- and γ-secretase enzymes. Cerebral parenchymal Aβ deposition can occur as diffuse plaques with little surrounding pathology or as fibrillar plaques associated with dystrophic neurons and inflammation. 20 The level of Aβ within the brain depends not only on the rate of Aβ production but also on the rate of its removal through various clearance pathways. An increasing number of strategies for treating AD are aimed at promoting the clearance of Aβ in the brain. The major aim of this study was to assess how the presence or absence of TLJN treatment affects the content of plasma Aβ and cerebral Aβ deposition in SAMP8 mice. For these studies, we examined cerebral Aβ expression with an Aβ antibody in each group of mice by immunohistochemical analysis. Then, the levels of plasma Aβ1-40 and Aβ1-42 were assessed by ELISA. In our studies, there was a significant elevation in the amount of total Aβ deposition in the hippocampus in model mice compared to control mice. In contrast to the model mice, a more striking reduction in the amount of cerebral Aβ was observed in the TLJN mice (Figure 2). These findings demonstrated that TLJN treatment in SAMP8 mice markedly affected the deposition of Aβ in brain. Moreover, these effects was consistent with that it also influenced the levels of plasma Aβ1-40 and Aβ1-42, as there was a significant elevation in the levels of plasma Aβ in model mice. In contrast to model mice, an obvious reduction in the plasma Aβ content was observed in TLJN mice (Figure 3). The system responsible for managing Aβ exchange between the brain and the periphery is regulating the pathways of Aβ periphery clearance, which include Aβ transport across the blood–brain barrier from brain to blood and Aβ perivascular lymphatic drainage to lymph nodes. First, TLJN influenced the deposition of Aβ in brain, then through improved Aβ periphery clearance, regulating the main transporters on the blood–brain barrier, or maintaining the function of peripheral tissues, hence to reduce the levels of plasma Aβ. Based on the above-mentioned data, TLJN could reduce the content of plasma Aβ, and also diminished cerebral Aβ deposition in SAMP8 mice.
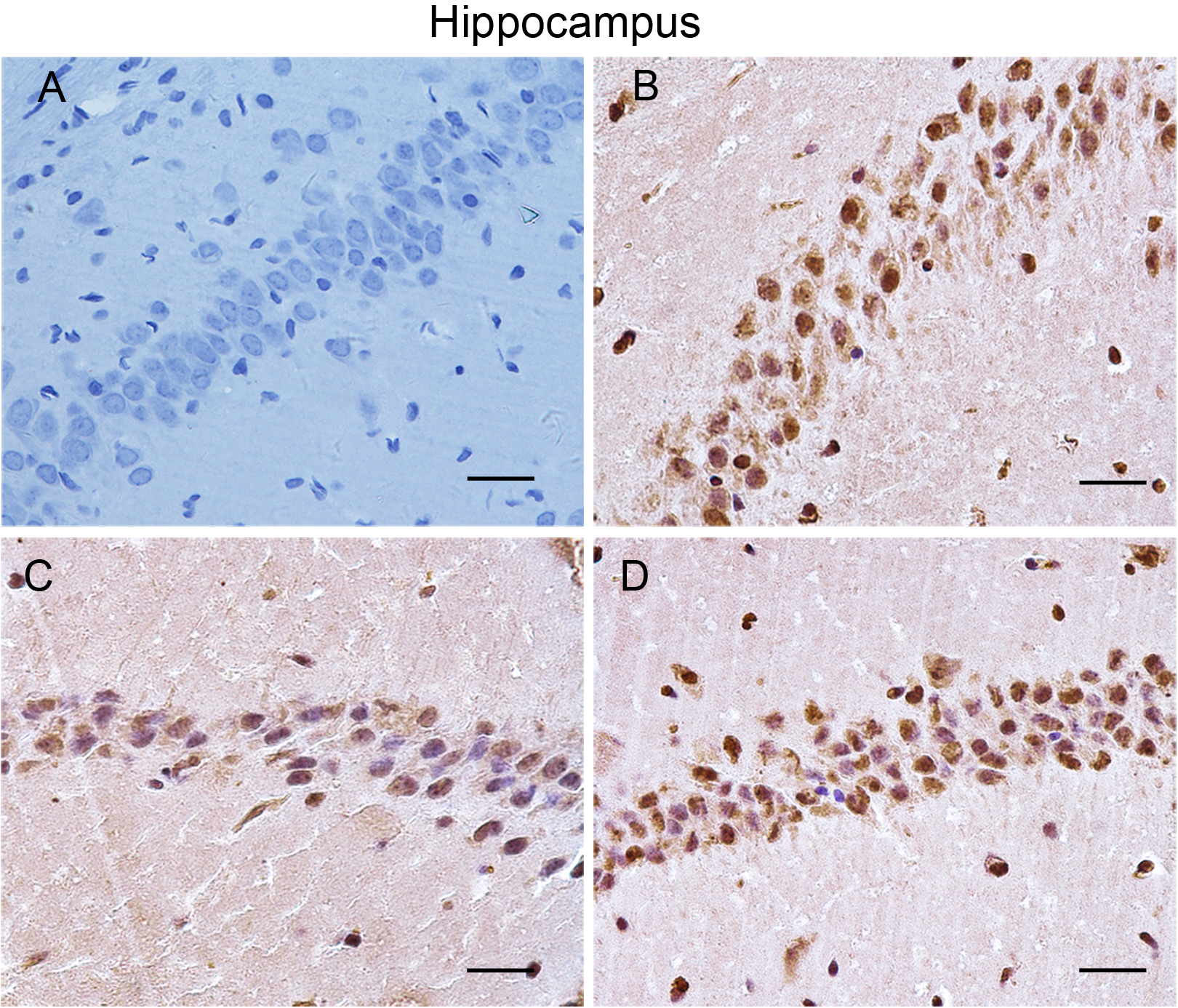
Cerebral amyloid β (Aβ) depositions. A–D, Representative sections from control (A), model (B), Tong Luo Jiu Nao (TLJN) (C), and donepezil hydrochloride (DNP; D) mice. Scale bar = 30 µm.
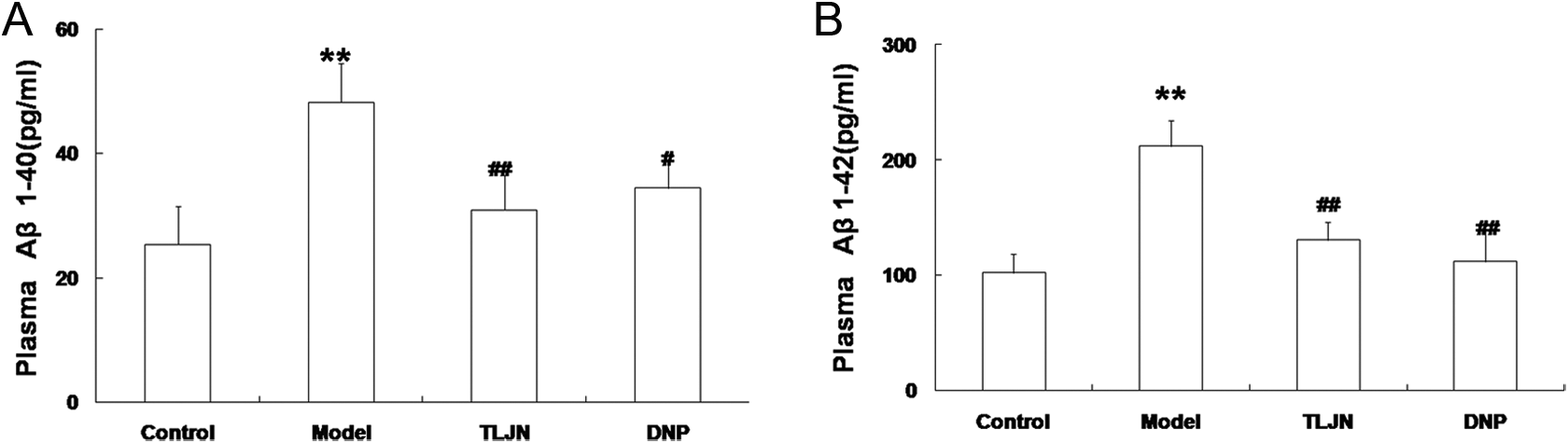
The levels of plasma Aβ. A and B, Quantification of plasma amyloid β (Aβ)1–40 (A) and Aβ1–42 (B). *P < .05, **P < .01 versus the control group. #P < .05, ##P < .01 versus the model group.
TLJN Reduces the Expression of Several Inflammatory Mediators—VCAM-1, MIP-1β, and CCR5—in SAMP8 Mice
Recent studies have indicated that neuroinflammation is associated with cerebral Aβ deposition, where it may contribute to the progressive dementia that characterizes AD. 4,21,22 Adhesion molecules and chemokines have been directly linked to neuroinflammation, and their dysfunction may influence the processing of Aβ. 23 –25 In our study, using immunohistochemistry and immunoblotting, VCAM-1 protein expression was significantly increased in the cerebral hippocampus and cortex of model mice, but this effect was reduced in TLJN mice. Accordingly, levels of VCAM-1 protein were significantly reduced in TLJN mice compared to model mice (Figure 4). Furthermore, the chemokine MIP-1β and its receptor CCR5 have been shown to be elevated in model mice compared to control mice; however, we found markedly reduced expression of MIP-1β and CCR5 proteins in TLJN mice compared to model mice (Figure 5). Together, these findings implicate VCAM-1, MIP-1β, and CCR5 in the biological and pathological processes that underlie AD, and TLJN may be linked to a pronounced reduction in associated neuroinflammation.
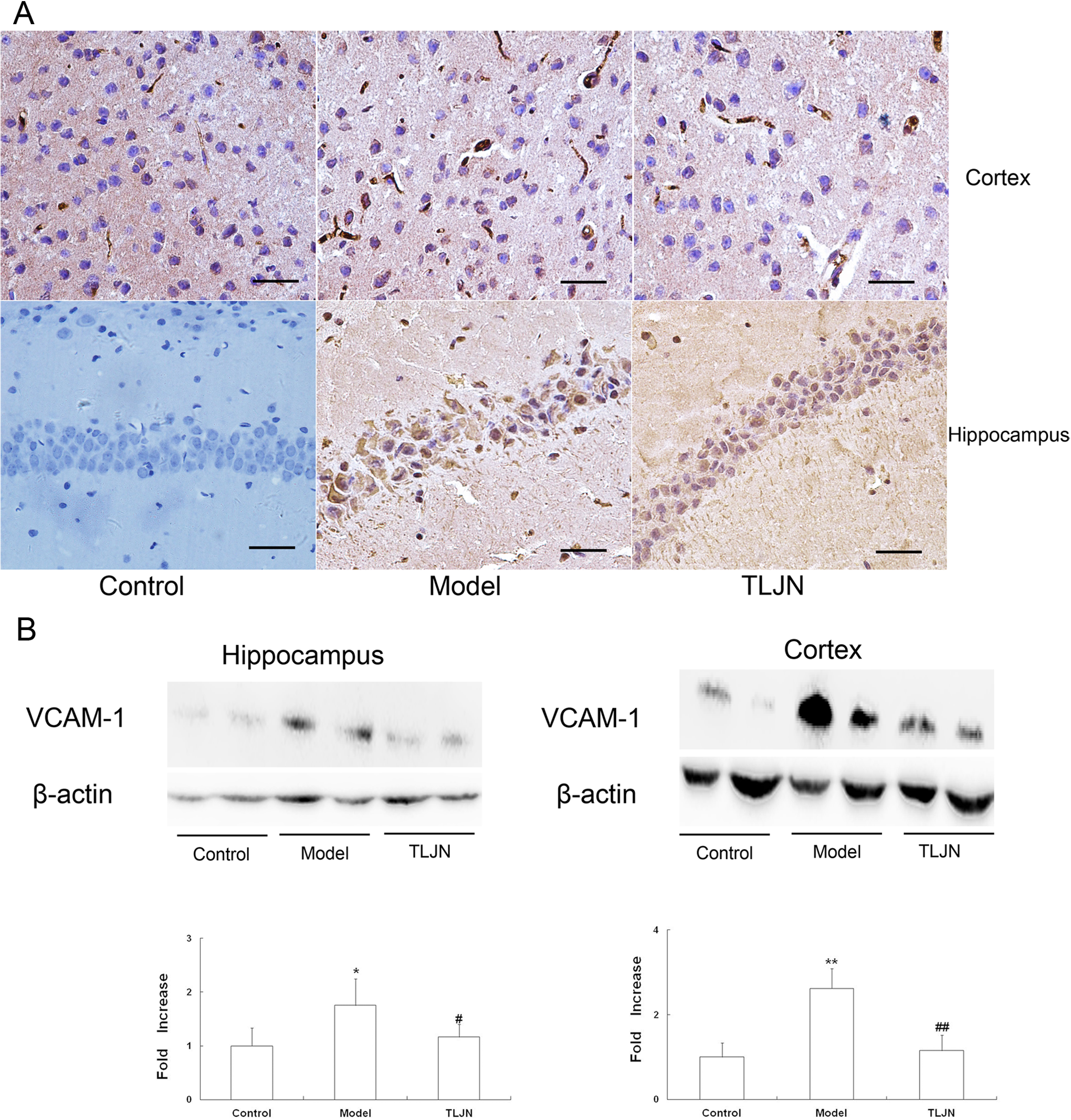
Expression of vascular cell adhesion molecule-1 (VCAM-1) in control, model, and Tong Luo Jiu Nao (TLJN) mice. A, Representative sections from control, model, and TLJN mice. Scale bar = 30 µm. B, Immunoblotting was performed to detect VCAM-1 expression in the hippocampus and cortex, followed by staining with anti-β-actin immunoglobulin G (IgG). *P < .05, **P < .01 versus the control group. #P < .05, ##P < .01 versus the model group.
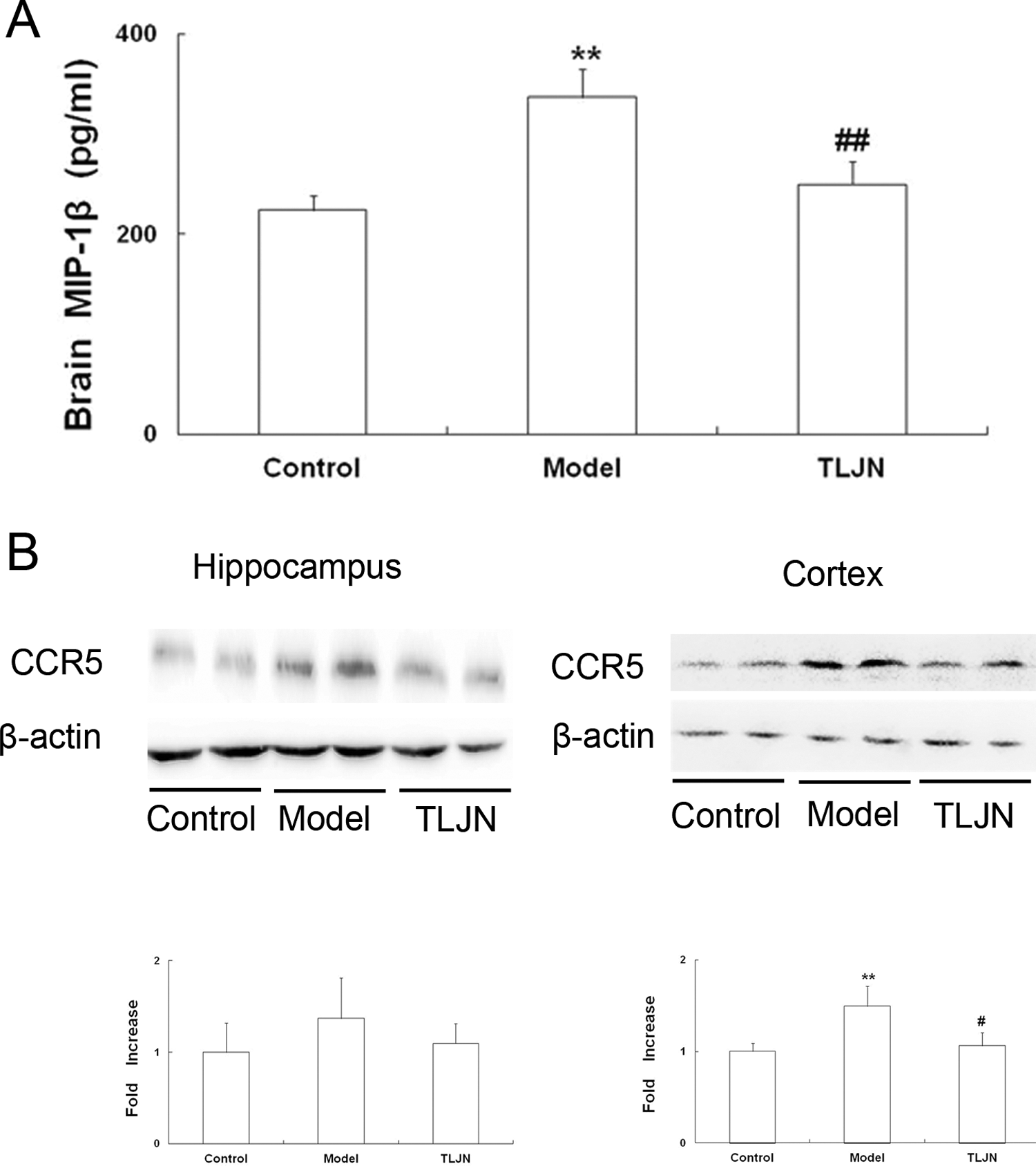
Expression of MIP-1β and its receptor, chemokine receptor 5 (CCR5), in control, model, and Tong Luo Jiu Nao (TLJN) mice. A, Levels of brain macrophage inflammatory protein-1β (MIP-1β). Enzyme-linked immunosorbent assay (ELISA) was performed to detect MIP-1β in brain tissues. B, CCR5 expression. Immunoblotting was performed to detect CCR5 expression in the hippocampus and cortex, followed by staining with anti-β-actin immunoglobulin G (IgG). *P < .05, **P < .01 versus the control group. #P < .05, ##P < .01 versus the model group.
Activation of JNK and ERK1/2 MAP Kinases
Using immunoblotting, we measured the phosphorylated forms of JNK and ERK1/2 in the cerebral hippocampus and cortex of control, model, and TLJN mice. Levels of phosphorylated JNK and ERK1/2 were significantly increased in brain extracts from model mice compared to control mice, whereas TLJN mice exhibited significantly less phosphorylation of JNK and ERK1/2 compared to model mice (Figure 6).
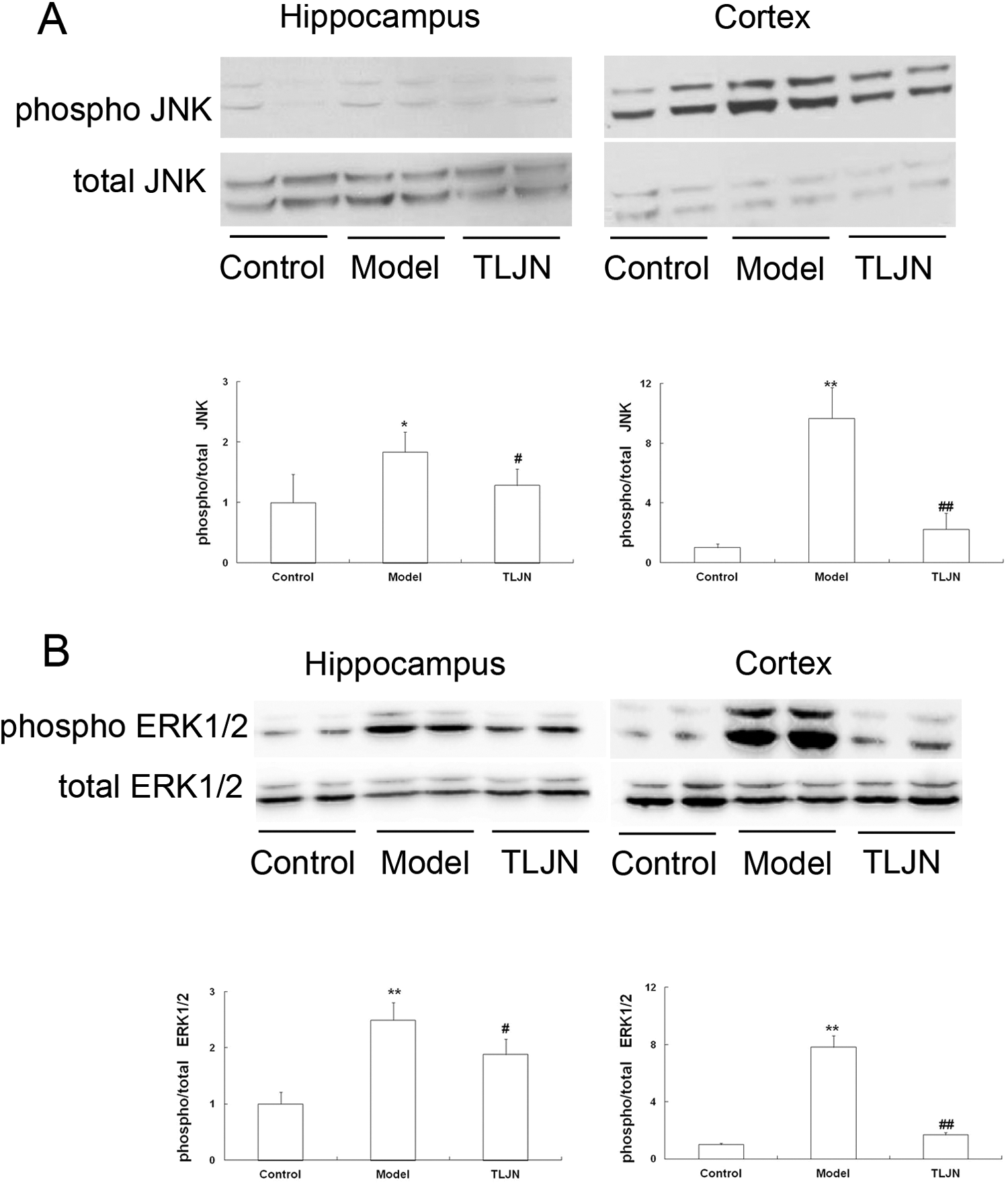
The effect of Tong Luo Jiu Nao (TLJN) on the phosphorylation of c-Jun N-terminal kinases (JNK) and extracellular Signal-regulated Kinase (ERK)1/2. The phosphorylation of JNK (A) and ERK1/2 (B) in brain homogenates of the indicated mice was measured by immunoblotting; the vertical axis indicates the fold increase in phosphorylated JNK and ERK1/2 (normalized to total JNK or ERK1/2, respectively) in the indicated mice compared to the control mice. *P < .05, **P < .01 versus the control group. #P < .05, ##P < .01 versus the model group.
Discussion
Alzheimer’s disease is characterized by the accumulation of toxic Aβ proteins in the extracellular space and on the blood vessel walls in the brain. 26,27 Inflammatory changes have also been frequently observed in the AD brain; however, these have traditionally been interpreted to be independent “copathologies” that may be further downstream and would confound rather than support the early detection and reliable diagnosis of AD. It is increasingly recognized that neuroinflammatory pathologies may characteristically or even specifically contribute to the development of amyloid pathology and/or the neurodegeneration and cognitive decline that occur in AD. 28,29 Additionally, many studies have shown the activation of inflammatory pathways in pathologically vulnerable regions of the AD brain and have documented the presence of many inflammatory mediators. 30,31
The role of inflammatory response in AD is topics of high current interest, and the most important morphological features of inflammatory response are inflammatory cell infiltration. In the process of inflammatory cell infiltration, the key points include adhesion and chemotaxis. The role of cellular adhesion molecules has been best characterized in leukocyte–endothelial cell interactions and subsequent transendothelial migration events. 32 Cellular adhesion molecule, VCAM-1, is principally expressed by endothelial cells and is involved in the adhesion of lymphocytes and monocytes, but neutrophils, to the endothelium. 33 It has been reported that VCAM-1 has been linked to the pathogenesis of AD. Zuliani and colleagues found that, compared to normal elderly control patients, plasma levels of VCAM-1 were increased in both patients with AD (n = 60) and those with vascular dementia (n = 80), independent of the presence of small- or large-vessel disease. 34 This finding is consistent with our observation that the expression levels of VCAM-1 were enhanced in the model group compared to the control group (Figure 4). Besides, chemokine MIP-1β and its specific receptor CCR5 also play causative roles in the pathophysiology of AD. 17,35 It was suggested that they are important regulatory molecules during cerebral inflammatory reactions in AD. To address this point, we first measured the levels of MIP-1β in the brains of each group and found that the levels of MIP-1β in the model group were markedly upregulated (P < .01). We then assessed the expression levels of CCR5 (Figure 5) and found that CCR5 expression was highly upregulated in the cortex of the model group (P < .01). Taken together, these findings indicated that the levels of VCAM-1, MIP-1β, and CCR5 protein were significantly increased in the model group. These data add to a growing literature that supports the importance of inflammatory mediators in the pathogenesis of AD.
TLJN, a modern Chinese medicine formula, is composed of geniposide, geniposidic acid, and ginsenoside Rg1. We previously showed that TLJN was efficacious in the treatment of brain diseases, such as AD. 9,11 In vitro, TLJN-treated brain microvascular endothelial cells exhibited downregulation of MIP-1β and CCR5 expression in a model of cerebral ischemic injury, suggesting a possible role for TLJN in cerebral vascular disease-associated inflammation. 17 Next, we sought to determine whether TLJN also had a direct role in reducing the inflammatory stresses that are associated with Aβ-induced neuroinflammation in AD. The role of TLJN as an inflammation repressor that mediates effects on an Aβ-rich environment is highlighted by our findings for the TLJN group. Our detailed mechanistic analyses linked these observations to the activation of JNK and ERK1/2 MAP kinases (Figure 6).
In this study, we have shown that TLJN reduced the content of plasma Aβ and cerebral Aβ deposition in an SAM model of AD. Moreover, TLJN reduced the levels of MIP-1β and downregulated the expression of VCAM-1 and CCR5 in both the hippocampus and the cortex and ameliorated signs of inflammatory stress in AD mouse brains. In the future, TLJN may have significant therapeutic potential and could possibly represent an alternative therapy for treating the pathological processes that underlie AD.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant no. 81303088).