
Abstract
As an endogenous cytoprotective factor, the protection of estrogen and heat shock protein-70 (Hsp70) on DNA has been documented, respectively, but the functional interaction between estrogen and Hsp70 on DNA damage repair is largely unknown. We therefore investigated the relation between estrogen and Hsp70 in terms of DNA protection in in vitro. The findings showed a significant reduction in cell survival and elevation in oxidative stress while cells were exposed to amyloid β (Aβ25-35) peptide, but preincubation of the cells with 17β-estradiol (17β-E2) ameliorated this situation. In addition, 17β-E2 alleviated oxidized DNA damage induced by Aβ and elevated the expression of Hsp70. However, the beneficial properties of 17β-E2 on reducing DNA damage were attenuated when Hsp70 gene was silenced accordingly. These results indicate that Hsp70 plays a role in DNA protection mediated by estrogen, and the DNA protection may be involved in Alzheimer’s disease preventive effect from estrogen.
Introduction
Excessive accumulation of amyloid β (Aβ) peptide in the brain is a major characteristic of Alzheimer’s disease (AD) and a critical cause of neurodegeneration. 1 Although the mechanism by which Aβ induces neurodegeneration remains controversial and unclear, there is evidence suggesting that oxidative damage plays a key role. 2 The Aβ peptides directly initiate free reactive oxygen species (ROS) formation 3 or induce ROS production in neurons through the activation of nicotinamide adenine dinucleotide phosphate oxidase 4 ; when the ROS production exceeds the capacity of detoxification, it can cause oxidative stress, which is widely recognized to damage the biological molecules including DNA. A substantial body of evidence indicates that oxidative DNA damage is a feature of AD in the brain as well as in peripheral tissues. Higher concentrations of oxidized pyridines and purines were detected in lymphocytes and leukocytes of patients with AD compared with controls, 5 and increased levels of oxidized DNA were observed in brains of patients with AD. 6–8 In particular, increasing evidence suggested that DNA damage, which occurred in cells exposed to Aβ may be a critical event in Aβ neurotoxicity. 9 , 10 Thus, prevention of such DNA damage or induction of DNA repair could improve the condition of patient with AD.
Heat shock protein-70 (Hsp70) is the major member of the 70-kDa heat shock protein family, and its expression is induced in elevated environmental temperatures and a wide variety of other stress, such as ultraviolet irradiation and oxidative stress. 11 The Hsp70, known as adenosine triphosphate-dependant molecular chaperones, maintains the normal functions of cells by binding to unfolded or misfolded proteins and promoting either correct refolding or proteolytic degradation of these proteins. 12 A few previous studies suggest that Hsp70 might be involved in DNA repair. For example, Abe et al 13 found that the nuclear translocation of Hsp70 may protect chromatin DNA from further damage or facilitate the repair of DNA damage. Xiao et al 14 reported that high Hsp70 levels generally correlated with lower DNA damage. Moreover, the increasing levels of cyclobutane pyrimidine dimers and 8-hydroxy-2′-deoxyguanosine induced by oxidative stress, both of which are DNA damage markers, can be suppressed in transgenic mice expressing Hsp70; these findings explicitly indicate that Hsp70 plays a role in DNA protection. 15 , 16
In addition to the classic role in reproduction, estrogen has an important role in maintaining neural functions. Although there are controversies regarding theuse estrogen, the benefits of estrogen supplementation in AD prevention against Aβ neurotoxicity in postmenopausal women has been confirmed by different research including the improvement in oxidative stress induced by Aβ through upregulation the expression of antioxidant enzyme. 17 , 18 Obviously, the presence of estrogen shall be beneficial to deal against DNA damage triggered by ROS. More importantly, accumulating studies have supported that estrogen deficiency supplementation may attenuate DNA damage, 19 , 20 suggesting that estrogen possesses the power to protect DNA. However, the mechanism of estrogen against DNA damage is still unelucidated. Previous studies reported that estrogen may facilitate Hsp70 expression in different cells under various stress conditions, 21 , 22 which make us postulate that the anti-DNA damage effect of Hsp70 may be involved in the beneficial effect from estrogen. To investigate the hypothesis, in the present study, Hsp70-silence cells were prepared, and the effect of estrogen on DNA damage was compared in the Hsp70-silence cells and the parental cells. The results partially explained the mechanism of estrogen DNA protective activity.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) and estrogen-free fetal bovine serum (FBS) were obtained from Gibco Invitrogen Corporation (Carlsbad, California). The reagent kits for DNA isolation were purchased from Qiagen (Qiagen GmbH, Germany). The protease inhibitor mixture and BCA Protein Assay Kit were purchased from Pierce Biotechnology (Rockford, Illinois). Oligofectamine transfection reagent was obtained from Invitrogen. The Hsp70 antibody (inducible form) was purchased from GeneTex (Irvine, California); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, California). The reagent kits for the measurement of malondialdehyde (MDA), total superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All other chemicals were obtained from Sigma-Aldrich (Saint Louis, Missouri).
Cell Culture and Treatment Protocols
Human neuroblastoma SH-SY5Y cells were obtained from the Cell Center, Institute of Basic Science, the Chinese Academy of Medical Sciences and grown in DMEM supplemented with 10% estrogen-free FBS, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin in a 5% CO2-humidified atmosphere at 37°C. 17β-estradiol (17β-E2) was dissolved in ethanol and added to cell culture media at designated concentrations with the final concentration of ethanol ≤0.1%. After 12 hours, the cells were exposed to Aβ25-35 for 24 hours. Then, the cells were collected for further research. The Aβ25-35 peptide stock solution of 1 mmol/L was prepared in sterile distilled water, stored at −20°C, and incubated for 72 hours at 37°C to aggregate before use.
Silencing of Hsp70 by RNA Interference
A 21-nucleotide-pair Hsp70 short interfering (si) DNA/RNA duplex (DNA-RNA hybrid) was chemically synthesized and purified by Shanghai Genechem (Shanghai, China). The DNA strand of the duplex was homologous to the Hsp70 DNA sequence: 5′-AAGGCCAACAAGATCACCAT-3′. The antisense RNA strand was 5′-AUGGUGAUCUUGUUGGCCU (dTdT)-3′. The control duplex carried 3 nucleotide substitutions (GCC to AAA in the DNA strand). The above DNA/RNA sequence followed the published data. 23 Transfection of cells with these siDNA/RNA duplexes was performed in 6-well plates with Oligofectamine according to the manufacturer’s protocol. Culture medium containing the transfection complexes was removed 12 hours after transfection, and the cells were then washed with phosphate-buffered saline (PBS) twice and cultured in regular medium for another 12 hours followed by 17β-E2 or Aβ treatment for further experiment.
Cell Viability Assay
Cells were plated in 96-well plates; after various treatments, cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The MTT was applied to the cultures at a final concentration of 0.5 mg/mL for 4 hours at 37°C. The medium was then aspirated, and 100 μL dimethyl sulfoxide (DMSO) was added to solubilize the colored formazan product. Absorbance was determined at 578 nm on a scanning multiwell plate reader (Dynatech Lab, Chantilly, Virginia, USA) after agitating the plates.
Measurement of MDA Content and Antioxidant Enzyme Activities
For assay of lipid peroxide and antioxidants, the cultures were washed with ice-cold PBS and then pooled in 0.1 mol/L PBS and 0.05 mmol/L EDTA-buffered solution and homogenized. The homogenate was centrifuged at 11 000g for 20 minutes at 4°C, after which the protein concentration was determined by the BCA Protein Assay Kit, using bovine serum albumin as a reference standard. The supernates were collected and stored at −80°C until use. The levels of MDA and antioxidant enzyme activities including SOD and GSH-Px were determined according to the instructions of the reagent kits.
Western Blot Analysis
Cells were lysed with radioimmunoprecipitation buffer (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]) supplemented with NaF and a protease inhibitor mixture. Typically, 50 µg of proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. The following primary antibodies were used: anti-Hsp70 (1:500) and anti-GAPDH (1:4000). Protein bands were detected using horseradish peroxidase-conjugated secondary antibodies and visualized using enhanced chemiluminescence. Scanned images of the developed blots were quantified using densitometry functions in Image-Pro Express 4.0 (Media Cybernetics Inc., Bethesda, Maryland, USA).
Single-Cell Gel Electrophoresis Assays
The single-cell gel electrophoresis (also termed comet assay) technique was performed as described previously with minor modifications. 24 In brief, slides were prepared with 85 μL of 0.5% normal melting agarose. The 1 × 10 4 cells were then mixed with 75 μL of 0.7% low-melting agarose and applied to the prepared slides. The slides were immersed in freshly prepared cold alkaline solution (2.5 mol/L NaCl, 100 mmol/L Na2EDTA, 10 mmol/L Tris, and pH 10) with 1% Triton X-100 and 10% DMSO for 1 hour. The slides were then placed in a horizontal gel electrophoresis chamber containing alkaline buffer solution with NaOH (10 mmol/L) and Na2EDTA (200 mmol/L) at pH 13.2. After a 20-minute DNA “unwinding” period, electrophoresis was performed at 25 V and 300 mA for 20 minutes. Following neutralization (Tris-HCl, pH 7.5), the slides were stained with propidium iodide (5 μg/mL) for 15 minutes and examined using fluorescence microscope (Olympus, Japan) with an excitation filter at 515–560 nm. Random fields were selected at a constant depth of the gel, avoiding the edges. The comet tail length, which is defined as the diameter of the nucleus plus the migration distance of DNA, was measured using CASP software (CASP-1.2.2, download in http://casp.sourceforge.net/index.Php). The DNA migration of 30 randomly selected cells was examined for each sample.
DNA Extraction and Detection of 8-Hydroxydeoxyguanosine (8-OHdG) Formation
The levels of 8-Oxo-2'-deoxyguanosine (8-OHdG) were determined using high-performance liquid chromatography (HPLC) method, as described before. 25 DNA was isolated from the SH-SY5Y cells using a commercial kit following the manufacturer’s instructions. The purity of the sample DNA was verified with a criterion of an A260-A280 ratio of 1.7: 1.8. The isolated DNA (50 μg) was digested to nucleotides with nuclease P1 and then treated with alkaline phosphatase. The hydrolyzed DNA mixture was filtered by a 0.45-μm filter, and 10 μL of extracts were analyzed using Waters HPLC system (Waters Corporation, Milford, Massachusetts, USA). A high-throughput, small particle, large-diameter column [Platinum EPS C18 column (Alltech Associates Inc. Deerfield, Illinois,USA); particle size 0.5 μm, 55-7 mm] was used to separate the analyte. The mobile phase, consisting of 125 mmol/L citric acid, 250 mmol/L sodium acetate, 100 mmol/L acetic acid, and 10% methanol, was run at a flow rate of 1.2 mL/min. The 8-OHdG and dG were measured simultaneously using a Waters 464 Pulsed Electrochemical Detector (Waters Corporation, Milford, Massachusetts, USA) and a Waters 996 Photodiode Array Detector(Waters Corporation, Milford, Massachusetts, USA). The retention time of dG was 3.2 minutes and 8-OHdG was 4.1 minutes. The amounts of 8-OHdG in the sample were calibrated with respective standards. The results were expressed as the number of 8-OHdG molecules per 105 dG.
Statistical Analysis
All experiments were repeated in triplicate. The results were expressed as mean ± standard error. Data were analyzed by Student t test and 1-way analysis of variance followed by post hoc multiple comparison tests, with P value less than .05 is considered to be statistically significant.
Results
17β-Estradiol Protected SH-SY5Y Cells Against Aβ25-35-Induced Cytotoxicity
To determine the toxicity of Aβ25-35 induced, we exposed the cultured SH-SY5Y cells to Aβ25-35 (0.1-30 μmol/L) for 24 hours, and the cell viability was assessed by the MTT reduction assay. The Aβ25-35 induced cell death in a dose-dependent manner, and the 50% inhibiting concentration (IC50) was 16.1 μmol/L (Figure 1A). Based on this result, 15 μmol/L was selected as the optimal Aβ25-35 concentration for the subsequent experiments. The cells were then treated with various concentrations (10−6, 10−7, 10−8, and 10−9 mol/L) of 17β-E2 for 12 hours, after which the cells were incubated with 15 μmol/L of Aβ25-35 for 24 hours. The cell viability was measured using an MTT assay. As shown in Figure 1B, pretreatment with 17β-E2 significantly increased the viability of cells against Aβ25-35-induced cytotoxicity. Since 10−7 mol/L 17β-E2 shows a suitable effect (the cell viability was 72%-75% of control), 10−7 mol/L was chosen for the further experiments.
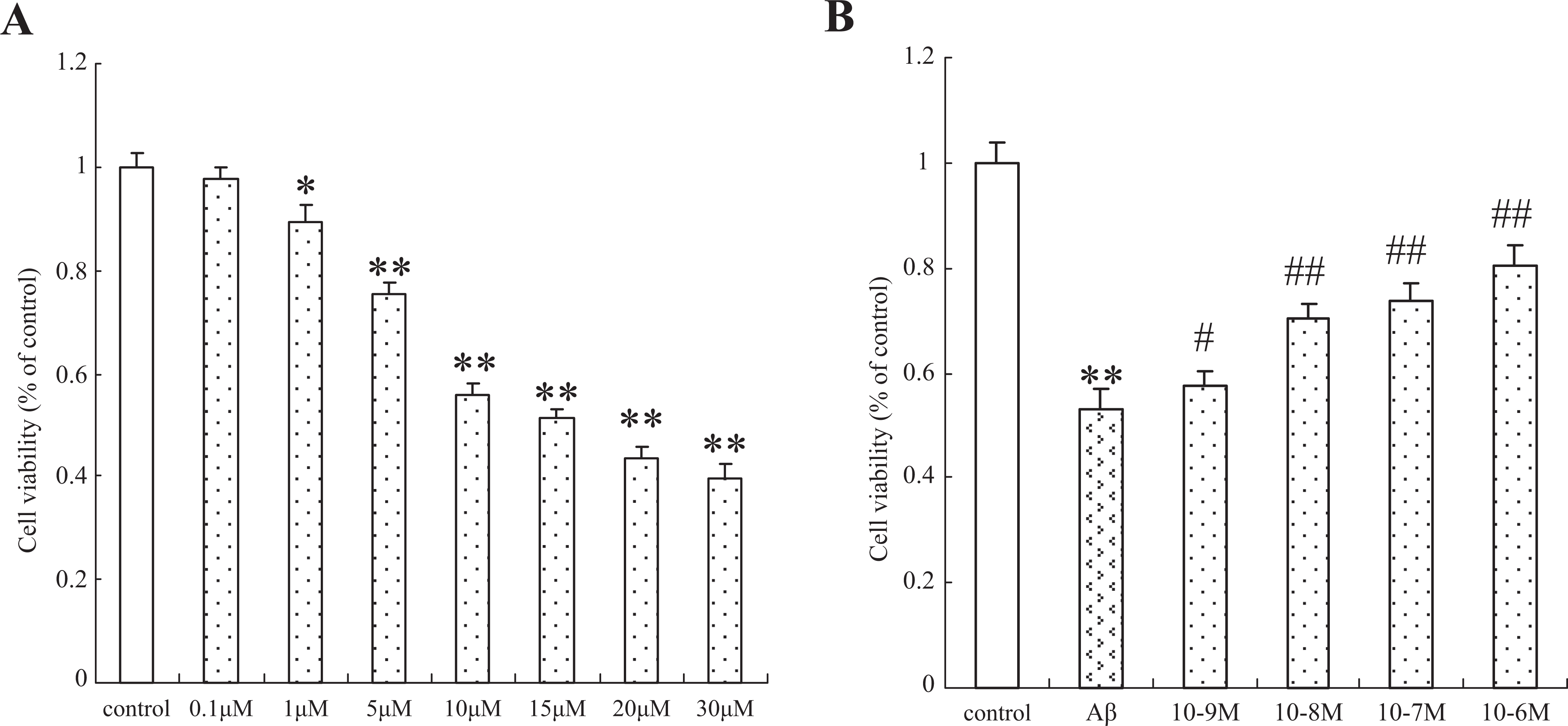
Protective effect of 17β-E2 on Aβ25-35-induced cytotoxicity in cultured SH-SY5Y cells. A, Cells were treated with the indicated concentrations (0.1-30 μmol/L) of Aβ25–35 for 24 hours. B, Cells were pretreated with different concentrations of 17β-E2 (10−9 to 10−6 mol/L) for 12 hours and then incubated with Aβ25-35 (15 μmol/L) for an additional 24 hours. Viability of cells was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Percentage of cell viability was relative to the untreated control cells. Values are represented as mean ± standard error of 3 independent experiments.*P < .05 and **P < .01 versus control group; #P < .05 and ##P < .01 versus Aβ25-35-treated cells. 17β-E2 indicates 17β-estradiol.
The Hsp70 RNA Interference Attenuated Neuroprotective Effect From 17β-E2
To analyze the role of Hsp70 in neuroprotective effect mediated by 17β-E2, we used the Hsp70 RNA interference (RNAi) approach. The cells were transfected with siDNA/RNA against Hsp70 or control siDNA/RNA. The specificity of Hsp70 siDNA/RNA was confirmed by the analysis of Hsp70 expression using Western blot, while it is hard to detect Hsp70 expression in interfere cells, and the level of Hsp70 was not affected in the control cells (Figure 2). And then, cell viability was evaluated using MTT assay. As shown in Figure 3, Hsp70 silence attenuated the effect of estrogen against cytotoxicity induced by Aβ. Although there was a decline in cell viability in RNAi control cells, the difference had no statistical significance when compared to normal control.
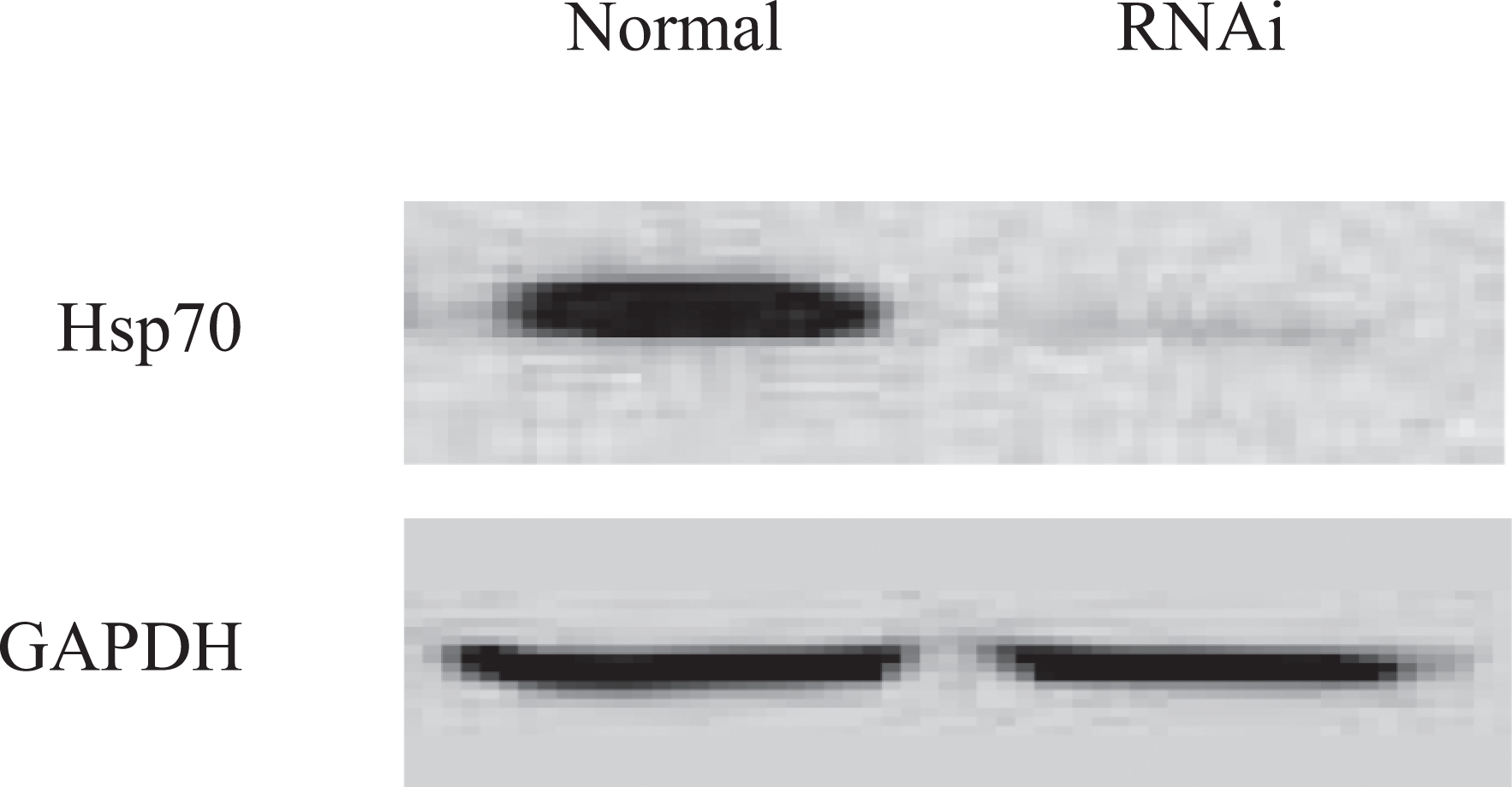
Cells were transfected with Hsp70-specific or control short interfering DNA/RNA duplexes, and then the expression of Hsp70 has been evaluated by Western blot 24 hours after infection. The levels of Hsp70 were decreased sharply in RNA interference cells but not been affected in normal cells. Hsp70 indicates heat shock protein-70.
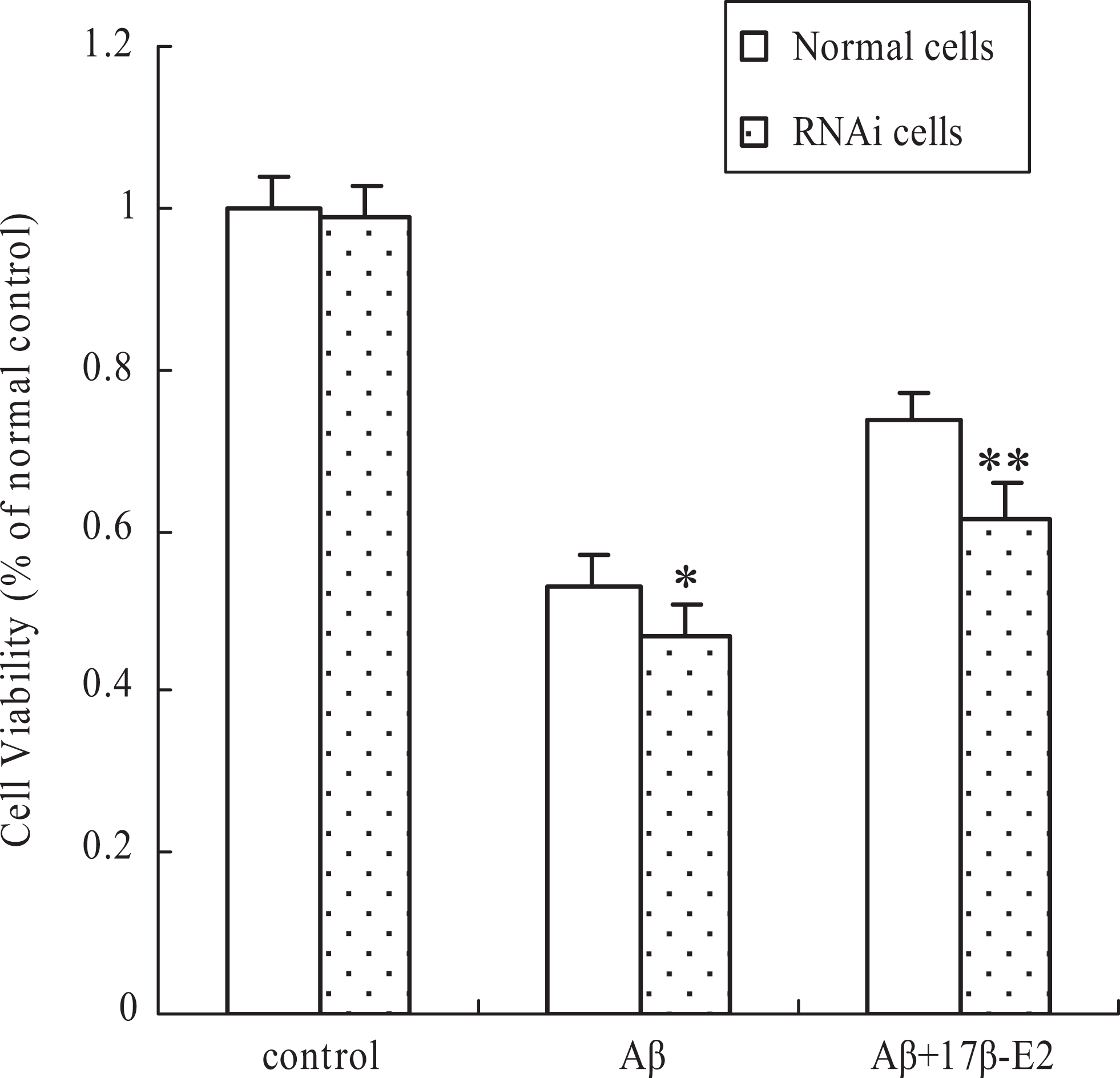
Heat shock protein-70 RNAi attenuated neuroprotective effect from 17β-E2. Normal or RNAi cells were pretreated with 17β-E2 for 12 hours and then incubated with Aβ25-35 for an additional 24 hours. Viability of cells was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Percentage of cell viability was relative to the normal control cells. Values are represented as mean ± standard error of 3 independent experiments. *P < .05 and **P < .01 versus normal cells. RNAi indicates RNA interference; 17β-E2, 17β-estradiol.
The Hsp70 RNAi Cannot Alter the Effect of 17β-E2 on Oxidative Stress
Oxidative stress plays an important role in the Aβ-induced neurotoxicity. Therefore, to examine the effect of estrogen on Aβ-induced oxidative stress, we measured the level of total SOD, GSH-Px, and MDA using a commercial assay kit. As shown in Table 1, incubation with Aβ25-35 for 24 hours significantly increased the contents of MDA (105.7%) in cells when compared to control. Moreover, preincubation with 17β-E2 attenuated the contents of MDA by 34.8% when compared to the cells treated only with Aβ25-35. In addition, exposure to Aβ25-35 significantly decreased the activities of SOD (51.2%) and GSH-Px (42.9%) in cells when compared to control. However, pretreatment with 17β-E2 showed a significant increase in the activities of SOD (56.9%) and GSH-Px (45.7%) when compared to the cells treated with Aβ25-35 alone. We also evaluated the effect of 17β-E2 on oxidative stress in Hsp70 RNAi cells; the results revealed the increase in MDA, but decrease in SOD and GSH-Px in Aβ25-35-treated cells and 17β-E2 may ameliorate these changes. Although the alterations of oxidation–antioxidation were striking in RNAi cells compared to normal cells, there were no significant differences in MDA and antioxidant enzyme between normal cells and RNAi cells (Table 1).
Effects of 17β-E2 on Lipid Peroxidation and Antioxidant Enzyme Activities in Aβ25-35-Treated Cultured SH-SY5Y Cells.a
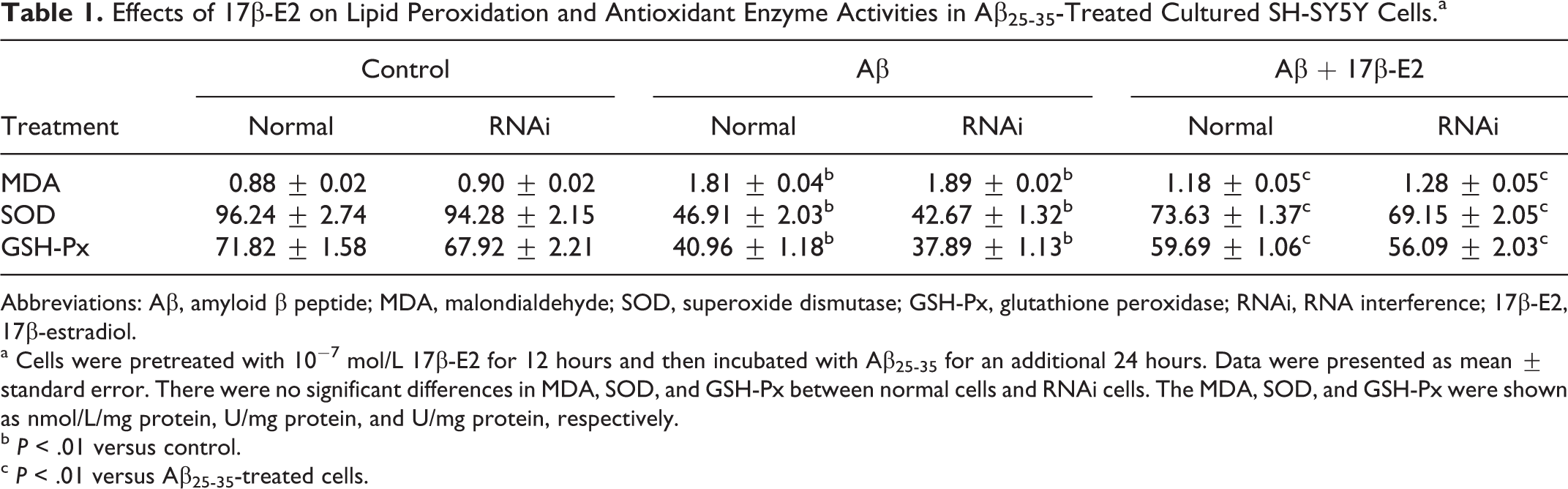
Abbreviations: Aβ, amyloid β peptide; MDA, malondialdehyde; SOD, superoxide dismutase; GSH-Px, glutathione peroxidase; RNAi, RNA interference; 17β-E2, 17β-estradiol.
a Cells were pretreated with 10−7 mol/L 17β-E2 for 12 hours and then incubated with Aβ25-35 for an additional 24 hours. Data were presented as mean ± standard error. There were no significant differences in MDA, SOD, and GSH-Px between normal cells and RNAi cells. The MDA, SOD, and GSH-Px were shown as nmol/L/mg protein, U/mg protein, and U/mg protein, respectively.
b P < .01 versus control.
c P < .01 versus Aβ25-35-treated cells.
The 17β-E2 Attenuates DNA Damage Induced by Aβ25-35
To investigate the effect of 17β-E2 on DNA damage, we first measured the DNA breakage in individual cells using comet assay. Cells with increased DNA damage display increased DNA migration from the nucleus toward the anode, which resembles the shape of a comet, and the length of the comet tail reflects the extent of DNA damage. As shown in Figure 4, although the cells treated with Aβ25-35 (including Aβ25-35 alone and Aβ25-35 plus 17β-E2 group) evidently showed comet tail, the comet tail is shorter in the cells cotreated with 17β-E2 compared to cells treated with Aβ25-35 alone (P < .01). We then assessed the level of 8-OHdG, a biomarker of DNA oxidation damage. The HPLC results showed that the level of 8-OHdG was sharply increased in the cells treated with Aβ25-35 (P < .01 compared to control cells); however, although cells received 17β-E2 before Aβ25-35, these changes were reversed and there was significant difference compared with Aβ25-35 group (Figure 5, P < .01). The above results indicated that the extent of DNA damage decreases while cells received estrogen.
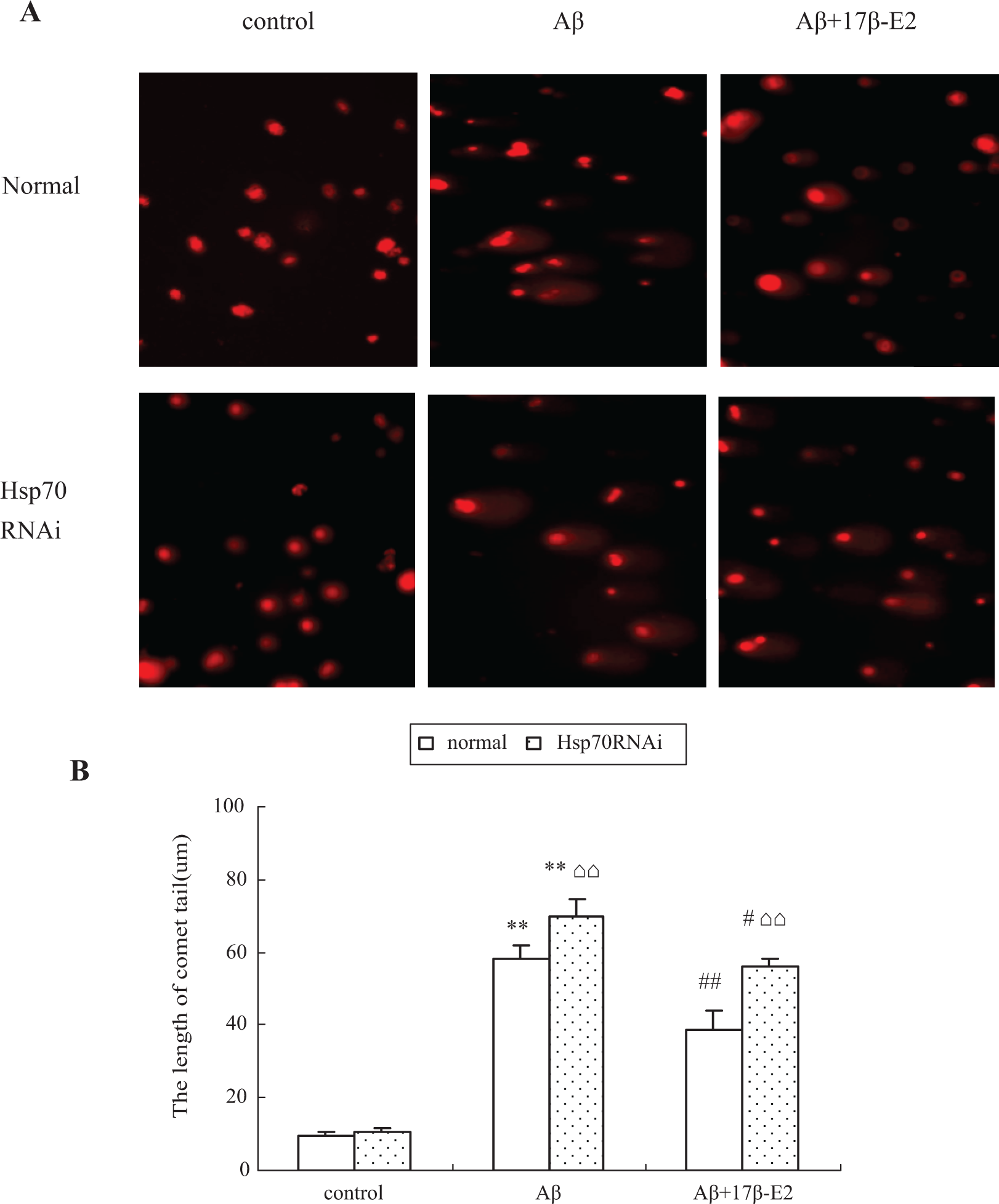
A, The comet image of the fluorescence photomicrographs showing DNA damage. The normal cells treated with Aβ25-35 showed evidently longer comet tail, and this situation can be alleviated while the cells cotreated with 17β-E2, but the DNA protective effect of 17β-E2 has been weakened by Heat shock protein-70 RNAi. B, Quantitative analysis of comet tail. *P < .05 and **P < .01 versus control group; #P < .05 and ##P < .01 versus Aβ group; ⌂⌂P < .01, RNAi cells versus normal cells in the same treatment. RNAi indicates RNA interference; 17β-E2, 17β-estradiol.
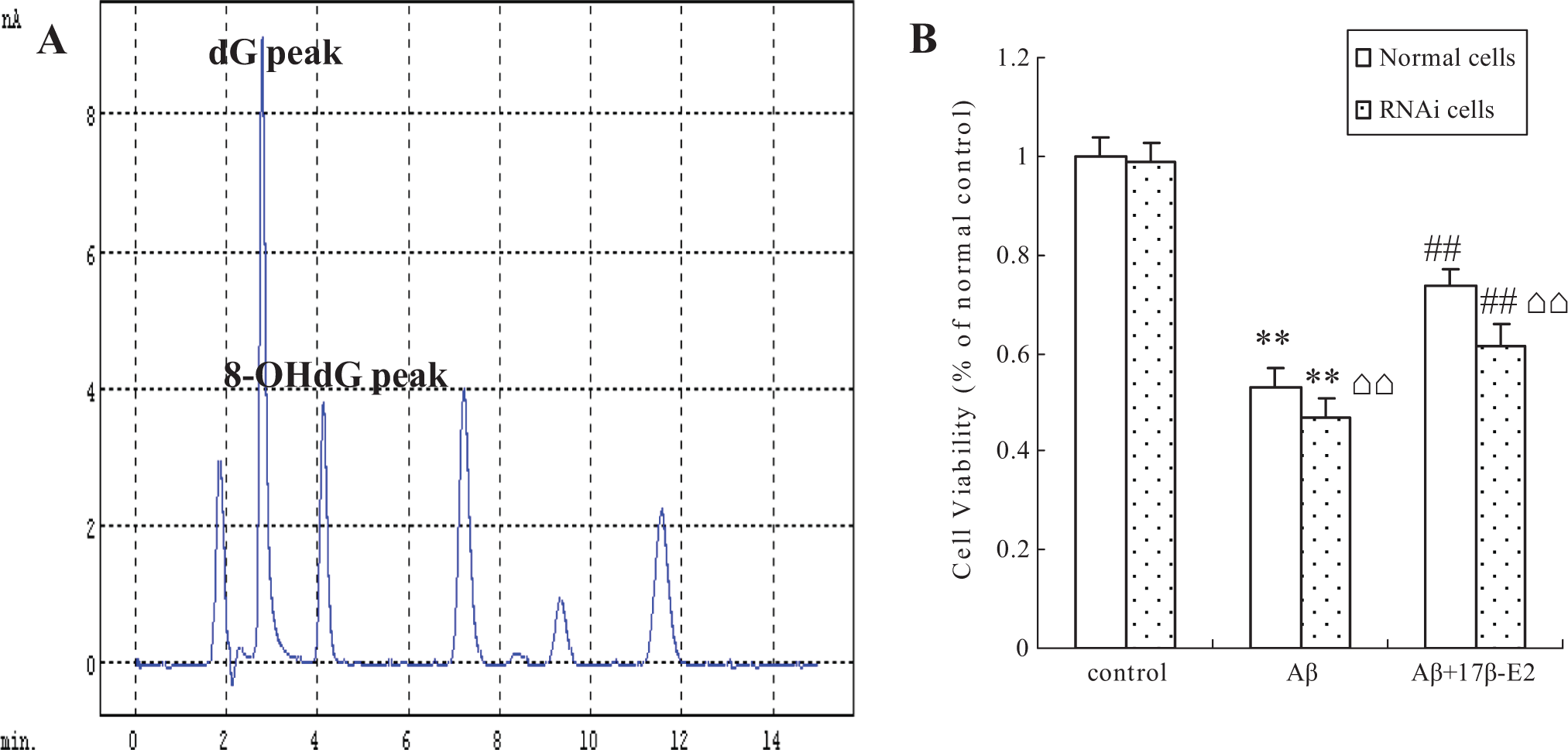
A, A typical HPLC chromatogram of sample analyte. The retention time for dG was 3.2 minutes, and 8-OHdG was 4.1 minutes. B, Quantitative analysis of 8-OHdG. *P < .05 and **P < .01 versus control group; #P < .05 and ##P < .01 versus Aβ group; ⌂⌂P < .01, RNA interference cells versus normal cells in the same treatment. 8-OHdG indicates 8-hydroxydeoxyguanosine; HPLC, high-performance liquid chromatography.
Suppression of Hsp70 Expression by RNAi Weakens DNA Protective Effect From 17β-E2
To analyze the potential involvement of Hsp70 in the anti-DNA damage properties of estrogen, we examined the expression of Hsp70 by Western blot. As shown in Figure 6, Aβ by itself increased Hsp70 expression compared to controls (P < .01). Moreover, the level of Hsp70 also elevated significantly when cells received 17β-E2 pretreatment before Aβ assault (P < .01 compared to Aβ alone group). The findings suggested that the involvement of Hsp70 in the mechanisms of estrogen mediates DNA protective effect. To verify this potential link, we evaluated the comet tail and 8-OHdG again in Hsp70 RNAi cells. It was observed that a rise in comet tail size and 8-OHdG ratio was present in the RNAi cells in comparison to those in the normal cells (Figures 4 and 5, respectively, P < .01). This result favorably suggested that Hsp70 is involved in the anti-DNA damage properties of estrogen.
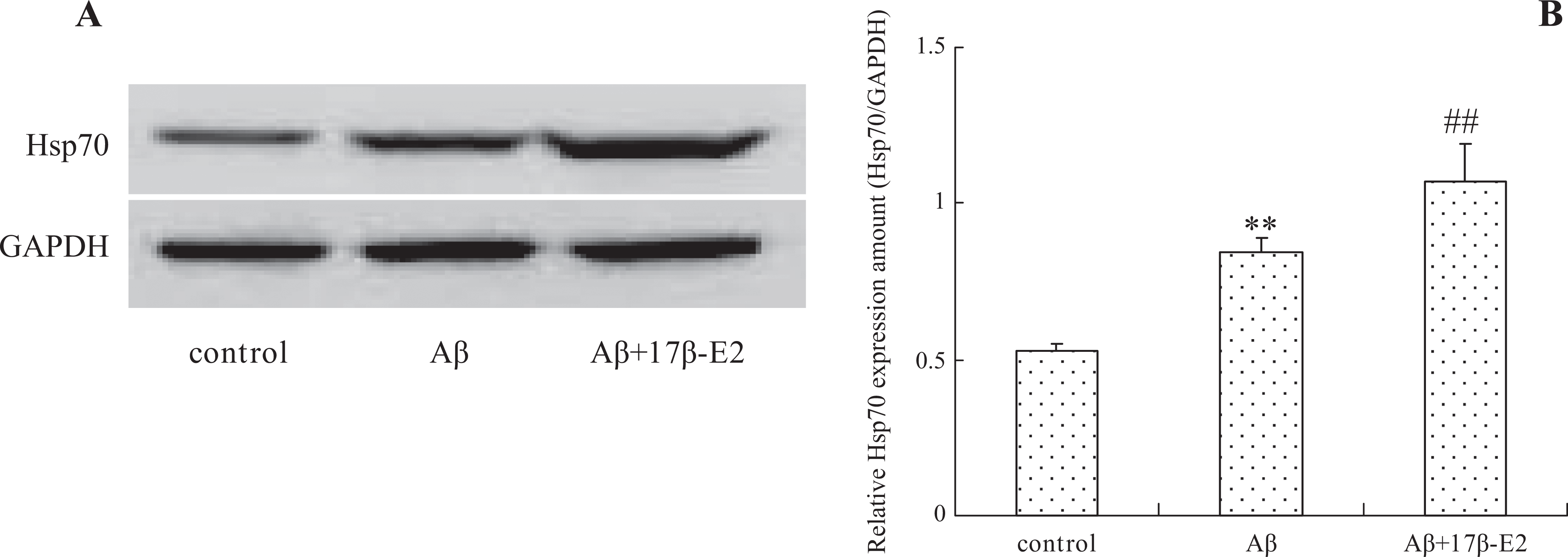
Effect of 17β-E2 on Hsp70 expression. A, Hsp70 expression has been induced by Aβ25-35 itself, and 17β-E2 treatment also facilitated Hsp70 expression indicated by Western blot experiments. B, Quantitative analysis of protein levels by densitometry. Densitometric analysis is mean ± standard error of 3 independent experiments. **P < .01 versus control group; ##P < .01 versus Aβ25-35 group. Hsp70 indicates heat shock protein-70; 17β-E2, 17β-estradiol.
Discussion
Estrogen is a well-known neuromodulatory and neuroprotective hormone, which is usually used in postmenopausal women for AD prevention. In this study, we showed that estrogen counteracts oxidized DNA damage induced by exposure to Aβ25-35 fragment in the SH-SY5Y cell line, but the beneficial properties of estrogen on improving DNA damage can be attenuated partially when the Hsp70 gene was silenced accordingly. The present finding revealed that estrogen offers a protective effect on DNA against Aβ and Hsp70 and plays a role in this process.
The Aβ is a soluble protein containing 40 to 42 amino acid residues that significantly contribute to the initiation and/or progression of neurodegenerative changes in AD. 26 Although amyloid proteins Aβ1-40 and Aβ1-42 are the major components of β-amyloid protein deposits in the AD brain, the structure–activity studies using Aβ fragments revealed that the peptide bearing the 11 amino acids (25-35) retains the ability to self-aggregate and mediates the toxicity in vitro. 27 In most experimental systems, the biological effects of Aβ1-40 and Aβ1-42 as well as the shorter fragment Aβ25-35 are comparable. 28
Although the mechanisms underlying Aβ toxicity are not clearly defined, there is a confluence of opinion suggesting that oxidative stress is mediated by ROS, which is deeply involved in the neurotoxic effects of Aβ, and along with the accumulation of excessive ROS, DNA oxidation will be induced, which recently has been considered as a trigger for AD. 29 Thus, modulation of oxidative stress and subsequently oxidative DNA damage induced by Aβ shall benefit AD prevention. To assess the effect of estrogen on oxidative stress, we chose to monitor the change in MDA, SOD, and GSH-Px. The MDA is an end product of lipid peroxidation and have been considered as a late biomarker of oxidative stress and cellular damage. Similar to our previous report, 17 the present study showed that Aβ25-35 caused a marked decrease in cell survival, which was accompanied by increase in the oxidative stress as shown by accumulation of MDA. However, the increase in cell death and MDA level induced by Aβ25-35 was significantly attenuated when the cells were pretreated with 17β-E2. The SOD and GSH-Px are well-known parameters like the antioxidative enzymes that react with ROS and neutralize them to alleviate oxidative stress. The decrease in SOD and GSH-Px has been shown in patients with AD 30 and is related to accelerating oxidative stress-induced neuronal damage. 31 Our findings showed that SOD and GSH-Px activity in the cells with Aβ25-35 treatment was significantly decreased when compared with that in the untreated cells, and the decreased activity was significantly increased by 17β-E2 pretreatment. We then evaluated the change in MDA and antioxidant enzyme activities in Hsp70 RNAi cells. Although the findings demonstrated an increase in MDA and a decrease in antioxidant enzyme activities in these cells, no statistically significant difference was detected between these alterations in the RNAi cells and normal cells. It has been confirmed that estrogen upregulates SOD and GSH-Px expression via the estrogen receptor pathway, 32 which may be the reason why the difference in antioxidant enzyme activities between Hsp70-silence cells and normal cells is not significant. Since the present findings showed that the cell viability decreased in the Hsp70-silence cells compared to normal cells, we speculated that there might be other mechanism beyond oxidation–reduction exacerbating Hsp70-RNAi cell damage.
In respond to oxidative stress, DNA receives attacks from oxyradical leading to strand breaks and formation of at least 20 modified bases oxidation adducts. Such DNA damage may finally induce cell dysfunction and even cell death. 8 We therefore evaluated DNA break using the comet assay and base oxidation using HPLC. This study disclosed a substantial amount of the nuclear DNA migrated as comet tails in cells exposed to Aβ, meanwhile, a high level of 8-OHdG has also been found, indicating DNA damages were induced. The cells hence respond to the DNA damages by activating complex enzymatic DNA repair mechanisms like base excision repair (BER), nucleotide excision repair, homologous recombination, and nonhomologous end-joining pathways. Although the details of the mechanism of Hsp70 effect on DNA repair remain to be studied, there was clear evidences indicating that BER was involved. 33 For example, Mendez et al reported that Hsp70 greatly stimulated DNA polymerase β to fill in the single-strand gaps in the BER process. 34 Kotoglou et al also found that Hsp70 might activate x-ray repair cross-complementing group 1 (XRCC1) during BER, which interacts with polynucleotide kinase poly(ADP-ribose) polymerases 1 and 2, DNA ligase 3a, and DNA polymerase β, and the combined capacities of XRCC1 to interact with the DNA repair enzymes and their DNA substrates allowed a more efficient repair process by holding the damaged DNA together. 35 Moreover, it was observed that Hsp70 might reduce the sensitivity of DNA to oxyradical attack 15 and resistance to oxyradical induced oxidative DNA damage. 36 Therefore, the induction of Hsp70 shall be beneficial to accelerate DNA repair and decrease the accumulation of oxidation DNA. Similar to these accumulating reports, 37 , 38 the present study found that the expression of Hsp70 increased in cells receiving Aβ alone compared to the controls, which has been considered as a cytoprotective response to against cytotoxicity mediated by Aβ. 38 Moreover, we also found that the level of Hsp70 is higher in cells treated with 17β-E2 plus Aβ compared to cells receiving Aβ alone. Although the existing evidence has indicated that estrogen may induce Hsp70 expression, 21 , 22 to make sure that the profit of estrogen on DNA is really from Hsp70 inducing, we measured the change in comet tail and 8-OHdG in Hsp70-RNAi cells and found that Hsp70 silence attenuated the DNA protective effect of 17β-E2. This finding supported that Hsp70 plays a role in the beneficial function of estrogen on DNA protection.
In conclusion, the present findings demonstrated that 17β-E2 may improve oxidative stress and DNA damage induced by Aβ, and the induction of Hsp70 may be an important mechanism for estrogen protection against DNA damage.
Footnotes
Authors’ Note
Yilong Dong and Yanmei Wang contributed equally to this work.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grant from the National 973 Fundamental Project of China (2010CB934002) and the Science and Technology project of Yunnan Provincial Science and Technology Department (2011FZ005).