
Abstract
Background
According to the 2020 global cancer statistics released by the International Agency for Research on Cancer (IARC), CRC ranks third in the global cancer incidence rate (10.0%) and second in mortality (9.4%), 1 with colon adenocarcinoma (COAD) being one of the most common diseases. 2 COAD originates from the glandular epithelial cells of the colon and usually undergoes a process from benign adenoma to malignant adenocarcinoma. 3 Most COAD patients are diagnosed with resectable tumors that can be treated with surgery. For advanced colorectal cancer, the primary treatment strategy is targeted therapy combined with chemotherapy, including oxaliplatin or irinotecan. 4 However, on the one hand, early-stage COAD may not be subtle and difficult to diagnose, on the other hand, there are still many side effects of the current first-line chemoradiotherapy for advanced COAD, such as gastrointestinal bleeding and immune system damage. 5 In recent years, the incidence of COAD has been increasing worldwide with a decreasing age of onset, which is associated with changes in dietary habits, including an increased intake of high-calorie, high-protein, and low-fiber foods. 6 Moreover, the high death rate of COAD is due to diagnosis at an advanced stage, inappropriate treatments, and distant metastases with poor prognosis. Clinical studies have shown that the 5-year survival rate for COAD patients is approximately 56%. Based on further analysis of different stages of COAD, only 8% to 13% of COAD patients with advanced COAD (stage III and IV) survive for more than 5 years. 7 Therefore, COAD represents a significant public health challenge, adversely affecting individuals’ health and quality of life. 8 It highlights the critical need for developing accurate prognostic models and identifying predictive molecular biomarkers, which are essential for improving the outcomes of COAD patients and enhancing their long-term quality of life. 9
The occurrence and progression of tumors is a complex multi-factor process, which is recognized to be affected by the activation of cancer genes, inactivation of tumor suppressor genes, abnormal apoptosis, and defective DNA damage repair. 10 Cancer development usually requires the accumulation of multiple driver mutations. Increased mutation rates from faulty DNA repair, replication errors, carcinogen exposure, or aging fuel cancer progression by elevating genetic mutation occurrence. As is well known, such cancer driver mutations, key for oncogenesis, are targets for anticancer drug development. However, the impact and significance of mitochondrial genomic mutations in cancer remain less understood. 11 In tumor cells, mitochondria is an important characteristic which affects cell death susceptibility, oxidative stress regulation, metabolism, and signaling. 12 Research shows mitochondrial metabolism and reactive oxygen species (ROS) generation can promote tumor progression and metastasis by activating Kras-induced anchor-independent growth through the ERK-MAPK signaling pathway. 13 Mitochondria transmit signals into the nucleus, a process that has been shown to allow mitochondria from non-cancer cell lines to reverse the oncogenic properties of tumor cells within the same nuclear background, thereby helping to tumor therapy effects. Meanwhile, Mendelian randomization analyses shows that there is a potential causal relationship between mitochondria-related genes and tumors, such as VARS2 with lung cancer, NSUN4 with breast and prostate cancer. 14
Various peptides and molecules, such as Bcl-2 and HSP90 ATPase targeting mitochondria, have been used for cancer therapy because of their effect on cell metabolism. 15 It is pivotal to screen novel mitochondria-related genes that are associated with the carcinogenesis of COAD as therapeutic and prognostic biomarkers. Some studies have been carried out to investigate the function of mitochondria-related genes in COAD.16,17 Nevertheless, studies on the function and efficacy of mitochondria-related genes in prognosticating COAD have been inadequate and the relationship between mitochondrial genes and COAD still remains largely unknown. 18 Therefore, the study aims to employ bioinformatics and molecular biology techniques for the identification of mitochondrial biomarkers associated with COAD. This endeavor seeks to establish an integrative COAD prognostic model based on nuclear mitochondria-related genes.
In this study, mitochondrial genes obtained from the UCSC Xena database were screened for COAD-related genes. Originally, based on TCGA and MITOMAP, differentially expressed-nuclear mitochondrial-related genes (DE-NMRGs) were constructed. Then, Select the optimized gene combination through cox regression analysis to establish a Nomogram model. Moreover, the correlation between important COAD-related genes in the model and the immune environment was further analyzed. Finally, molecular biology techniques were used to verify nuclear-mitochondrial-related genes. The survival prediction model containing 9 nuclear mitochondria-related genes provides evidence for improving the early diagnosis and treatment rate and survival rate of patients with COAD.
Methods
Data
The gene expression data of COAD was downloaded from the UCSC Xena database (https://xenabrowser.net/datapages/), which was measured by the Illumina HiSeq 2000 RNA Sequencing platform and normalized by log2 (FPKM + 1). The original data contained 512 samples, including normal controls and COAD tumor samples. By matching the clinical information of the samples, 41 normal control (CTRL) samples and 438 COAD tumor samples with complete prognostic information were selected, totaling 479 samples. This dataset was set as the training dataset.
The GSE39582 dataset 19 was downloaded from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/), 20 which contained 585 samples, of which 519 COAD tumor samples with clinical survival prognostic information. The dataset was measured by the GPL570 Affymetrix Human Genome U133 Plus 2.0 Array platform. The dataset was used as the validation dataset.
Screening of Significantly Differentially Expressed-Nuclear Mitochondrial-Related Genes
Based on the Cancer Genome Atlas (TCGA) expression data, the differentially expressed genes (DEGs) between COAD versus CTRL were studied using Limma (Version 3.34.7, https://bioconductor.org/packages/release/bioc/html/limma.html). 21 The thresholds were FDR < 0.05 and |log2fold change (FC)|>1.
Then, the nuclear mitochondrial-related genes (NMRGs) from the Human Mitochondrial Genome Database (MITOMAP, https://www.mitomap.org/) 22 were downloaded and compared with the screened DEGs. The overlapping DEGs were preserved and defined as differentially expressed NMRGs (DE-NMRGs). Finally, the Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway enrichment analysis was performed on the retained DE-NMRGs with a p-value < 0.05 using DAVID (Version 6.8, https://david.ncifcrf.gov/).23,24
Construction of DE-NMRGs-Related Risk Prognostic Model
Along with the clinical and prognostic features from TCGA dataset, the significant DE-NMRGs related to survival prognosis were identified by the Univariate Cox regression analysis using the R3.4.1 language survival package (http://bioconductor.org/packages/survivalr/). 25 A p-value < 0.05 was used as the threshold for significant correlation.
Then, the NMRGs were subjected to survival regression analysis using the LASSO algorithm in the R3.6.1 language lars package (v1.2, https://cran.r-project.org/web/packages/lars/index.html) 26 to screen the optimized DE-NMRGs combination.
Based on the LASSO prognosis coefficient and the expression level of genes, the risk score (RS) model was constructed:

The RS values were calculated in both of the two datasets, and then the samples were grouped into two risk groups: High (RS ≥ median value) and Low (RS < median value). The interrelationship between each group and the survival prognosis was assessed using the Kaplan–Meier curve method of the survival package. 25
Construction of Prognostic Nomogram for Survival Rate
In the training dataset, the clinical factor distributions in the two groups were compared by Fisher's exact test, and then the independent survival prognostic clinical factors were recognized using the Cox regression analysis in survival package. 25 A p-value < 0.05 was used as the threshold.
In order to further study the independent prognostic factors, the rms package (Version 5.1-2, https://cran.r-project.org/web/packages/rms/index.html) 27 in R was used to construct nomogram models for forecasting survival rates at 1, 3, and 5 years. The C-index coefficient of the nomogram prognostic model was then calculated by R3.6.1 survcomp (Version 1.34.0, http://www.bioconductor.org/packages/release/bioc/html/survcomp.html). 28
Characteristic DE-NMRGs-Related Gene Mutation Analysis
The COAD sample mutation annotation format (MAF) files from TCGA database (https://gdc-portal.nci.nih.gov/) were downloaded, and maftools package (Version 2.6.05, https://bioconductor.org/packages/release/bioc/html/maftools.html) 29 was used to determine the mutation characteristics of DE-NMRGs in COAD samples.
Immune-Related Analysis
The proportion of various immune cell in TCGA database was analyzed with the CIBERSORT (https://cibersort.stanford.edu/index.php), 30 and the differences in the distribution of diverse immune cells were compared. The ESTIMATE/immune/stromal score and tumor purity of COAD tumor samples were calculated by R3.6.1 estimate package (http://127.0.0.1:29606/library/estimate/html/estimateScore.html). 31 Finally, the correlation between the characteristic DE-NMRGs and the immune cells and estimate scores was analyzed by the R3.6.1 cor function.
KEGG Enrichment Analysis Related to Risk Grouping
The KEGG signaling pathway significantly related to risk grouping was identified with Gene Set Enrichment Analysis (GSEA, http://software.broadinstitute.org/gsea/downloads.jsp) 32 with a p-value < 0.05.
Mining of Gene and Protein Product Detection Expression Levels in the HPA Database
The Human Protein Atlas (HPA) database (https://www.proteinatlas.org/) 33 was employed to analyze the protein immunohistochemical morphology of the screened important DE-NMRGs and protein products in diseased and normal tissues.
Experimental Verification
The mouse colon cancer cell line HCT116 was provided from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Cells were cultured in Dulbecco's-modified Eagle medium (Invitrogen Gibco, CA, USA) with 10% fetal bovine serum (FBS, BI, Beijing, China) and 1% penicillin–streptomycin (Invitrogen Gibco, CA, USA) in an incubator at 37 °C with 5% CO2. Four-week-old male BALB/c nude mice were carried out by the National Guidelines for the Use of Animal and housed in an SPF environment with an ambient temperature of 23 to 26 °C and a relative humidity of 40% to 60%. The research protocol was passed the animal ethics review (No:SH9H-2021-A249-1). ETHE1 (NBP3-00355, 1:1000) and LARS2 (NBP2-97497, 1:1000) were obtained from Novus Biologicals. LRPPRC (21175-1-AP, 1:5000) and GAPDH (10494-1-AP, 1:5000) were purchased from Proteintech. HRP-labeled secondary antibody (ab205718, 1:5000) was obtained from abcam.
Experimental Model Animal
An experimental liver metastasis model was established by injecting 2*106 HCT-116 cell suspensions into the splenic vein of nude mice. The mice were randomly divided into blank group (B, PBS injection group, n = 3), liver metastasis model group (M, treatment with PBS (0.1 ml each) by gavage, n = 3), drug group (D, treatment with Xeloda medication, 90 mg/kg (0.1 ml each) by gavage, n = 3). The mice were subjected to the corresponding treatment every 2 days. During the experiment, the growth and behavior changes of nude mice were observed regularly. Five to seven weeks after modeling, all group mice were anesthetized with 3% isoflurane. Finally, liver was collected and immersed in 50 ml centrifuge tubes containing formalin.
Western Blot Assay
Homogenization of liver metastatic were lysed in 1× PIPA buffer solution (containing protease inhibitor and phosphatase inhibitor) and placed on ice for 30 minutes. A Bradford protein assay reagent determined the protein concentration of the lysates (Bio-Rad, CA, USA). Twenty micrograms of the protein was injected into SDS-PAGE gel for electrophoresis for protein isolation. After electrophoresis, the proteins on SDS-PAGE were transferred to polyvinylidene fluoride (PVDF) membrane, which was then sealed with a 5% skimmed milk sealant at room temperature 1 hour. After TBST washing, the PVDF membrane and primary antibody were incubated overnight at 4 °C. On the second day, the membrane was washed with TBST for three3 times, and then incubated with HRP-labeled secondary antibody at 37 °C for 1 hour. Finally, the Ultra Signal Hypersensitive ECL chemiluminescence substrate (4 A Biotech Co., Ltd, Beijing, China) was used to develop the image, and the Chemidoc MP imaging system (Bio-Rad, California, USA). The protein band strength was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and the relative expression levels of the target proteins and the reference protein in GAPDH were calculated.
Statistical Analysis
All statistical analyses were conducted in R software (3.6.1) and GraphPad Prism 9.5.0 software was used to identify DEGs. The rms was used to construct nomogram models for forecasting survival rates at 1, 3, and 5 years. Estimate was used to calculate the ESTIMATE score, immune score, stroma score, and tumor purity. The cor function was used to analyze the correlation between the characteristic DE-NMRGs and the immune cells and estimate scores. Continuous variables were expressed as mean ± standard deviation. When the data met the criteria of normal distribution and homogeneity of variance, One-way analysis of variance (ANOVA) was employed; if not met, non-parametric test was used. p < 0.05 represented a significant difference.
Results
Identification and Enrichment Annotation of DE-NMRGs
A total of 1499 DEGs were obtained from the TCGA, and 651 DEGs were upregulated and 848 DEGs were downregulated (Figure 1A). A total of 62 overlapping genes of DEGs and NMRGs were defined as DE-NMRGs (Figure 1B). The GO function and KEGG signaling pathway analysis was carried out on 62 overlapping DE-NMRGs, and a total of 12 biological processes and 7 KEGG signaling pathways were obtained (Figure 1C, D). As shown in Figure 1C, the GO term of biological process including mitochondrial respiratory chain complex I assembly, tRNA aminoacylation for protein translation, and mitochondrial electron transport, NADH to ubiquinone were detected. ccording to the KEGG pathway analysis (Figure 1D), Aminoacyl−tRNA biosynthesis, Metabolic pathways, and Oxidative phosphorylation were mostly related to the DE-NMRGs.

Identification and enrichment analysis of differentially expressed-nuclear mitochondrial-related genes (DE-NMRGs). (A) Volcano plot of differentially expressed genes. (B) A Venn-diagram of the DEGs and nuclear mitochondrial-related genes to obtain DE-NMRGs. The top 10 Gene Ontology (GO) function terms (C) and Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathways (D) enriched by the overlapping differentially expressed-nuclear mitochondrial-related genes (DE-NMRGs). The horizontal axis represents the number of genes, and the vertical axis represents the term. The color indicates significance, and the size of the dot is proportional to the number of genes.
Prognostic Model of DE-NMRGs
To further explore the potential prognostic value of DE-NMRGs, univariate cox survival analysis was first applied to find out their prognostic role, and 19 DE-NMRGs were found to be associated with the prognosis of COAD patients (Figure 2A). The optimal combination of DE-NMRGs was further screened using the LASSO algorithm, and 10 optimal DE-NMRGs were finally obtained including leucyl-TRNA synthetase 2, mitochondrial (LARS2), prolyl-TRNA synthetase 2, mitochondrial (PARS2), ETHE1 persulfide dioxygenase (ETHE1), leucine rich pentatricopeptide repeat containing (LRPPRC), transmembrane protein 70 (TMEM70), alanyl-TRNA synthetase 2, mitochondrial (AARS2), SPG7 matrix AAA peptidase subunit, paraplegin (SPG7), Acyl-CoA dehydrogenase family member 9 (ACAD9), valyl-TRNA synthetase 2, mitochondrial (VARS2), and ATPase phospholipid transporting 8A2 (ATP8A2). (Figure 2B). Then, based on the median expression of these 10 DE-NMRGs, the samples were divided into high- and low-expression groups, and the correlation between expression levels and survival prognosis was analyzed using Kaplan–Meier (KM) curves. As shown in Figure 3, low expression levels of AARS2, ACAD9, ATPBA2, SPG7, and VARS2 had higher overall survival time, while high expression levels of ETHE1, LARS2, LRPPRC, PARS2 and TMEM70 had higher overall survival time. The RS formula was then constructed based on the LASSO regression coefficients of these 10 DE-NMRGs and their expression levels in the dataset:


(A) The process of building the signature containing 19 DE-NMRGs most correlated with prognosis. The hazard ratios (HRs), 95% confidence intervals (CIs) calculated by univariate Cox regression. (B) LASSO logistic regression analysis for selection of optimal DE-NMRGs from DE-NMRGs. (C) The Kaplan–Meier (KM) survival curve of the 10 optimized DE-NMRGs combination. The blue and red curves represent the low- and high-expression groups, respectively.

The Kaplan–Meier (KM) curve, risk score (RS) value distribution, and receiver operating characteristics (ROC) curve of the training dataset (A–C) and the validation dataset (D–F): (A and D) the blue and red curves represent the low-risk and high-risk sample groups, respectively; (B and E) survival time status; (C and F) the numbers in parentheses represent the specificity and sensitivity value for the corresponding ROC curves.
The risk score of each patient in TCGA database and GSE3958 dataset was calculated according to the RS formula, and the patients were then categorized into low-risk and high-risk groups based on the median value. The Kaplan–Meier survival analysis revealed that the Survival time in the low-risk group was significantly higher than that of the high-risk group in the training dataset (Figure 3A). Besides, the number of deaths was significantly higher in the high-risk group (Figure 3B). In the training dataset, the 1-, 3-, and 5-year RS area under the curve (AUC) value was 0.906, 0.900, and 0.899, respectively (Figure 3C), indicating the risk model had a good predictive efficiency. Figure 3D and E showed that significant differences were observed between the high and low risk groups, and the mortality rate increased with the risk score in the validation dataset. The RS AUC values for 1, 3, and 5 years were 0.872, 0.819, and 0.802, respectively, in the validation set (Figure 3F). These results demonstrate the good performance of the prognostic model in both the training and validation sets.
Nomogram Model for Independent Prognostic Factors
In the TCGA training dataset, the Fishers’ exact test was utilized to compare the variability of the distribution of each clinical factor across risk subgroups, and it was found that the distributions of Pathologic N, T, stage, and Tumor recurrence were significantly different across subgroups (Table 1). Then, univariate and multivariate Cox regression analyses based on training set showed that age (p = 0.0011), pathologic T (p = 0.0018), tumor recurrence (p = 0.045), and RS model status (p = 0.019) were considered independent prognostic factors (p < 0.05, Figure 4A).

The prognostic forest plots (A), nomogram survival rate model (B), and the survival nomograms predicting the 1-, 3-, and 5-year survival rate for independent prognostic clinical factors. (C) The horizontal axis represents the predicted survival, and the vertical axis represents the actual survival.
Comparison of Each Clinical Factor in Two Risk Groups

RS: risk score.
To further analyze the correlation between age, pathologic T, tumor recurrence, and RS model status and prognosis, the nomogram model was constructed (Figure 4B). As shown in Figure 4C, the calibration curve suggested that the nomogram-predicted survival rate was similar to the actual the actual 1-, 3-, and 5-year survival rates survival rates.
Characteristic DE-NMRGs Mutations
Somatic mutation data of COAD patients from the TCGA database were downloaded and visualized using the “maftools” package. Missense mutations were the most prevalent among COAD patients, and single nucleotide polymorphisms (SNPs) outnumbered deletions (DELs) or insertions (INSs). C > T was the dominant mutation type detected (Figure 5A). Figure 5A also displays the number of altered bases for each mutation type in each sample. In each mutation type, the number of the altered bases in different samples was listed in Figure 5A. Next, the top 10 mutated genes, including ATP8A2, SPG7, AARS2, LRPPRC, LARS2, ACAD9, VARS2, PARS2, TMEM70, and ETHE1, are shown in Figure 5A. A waterfall plot was generated to show the detailed mutation information in each sample, with different colors indicating different mutation types (Figure 5B). Figure 5C reveals that LRPPRC and AARS2, as well as ETHE1 and VARS2 had the highest correlation.
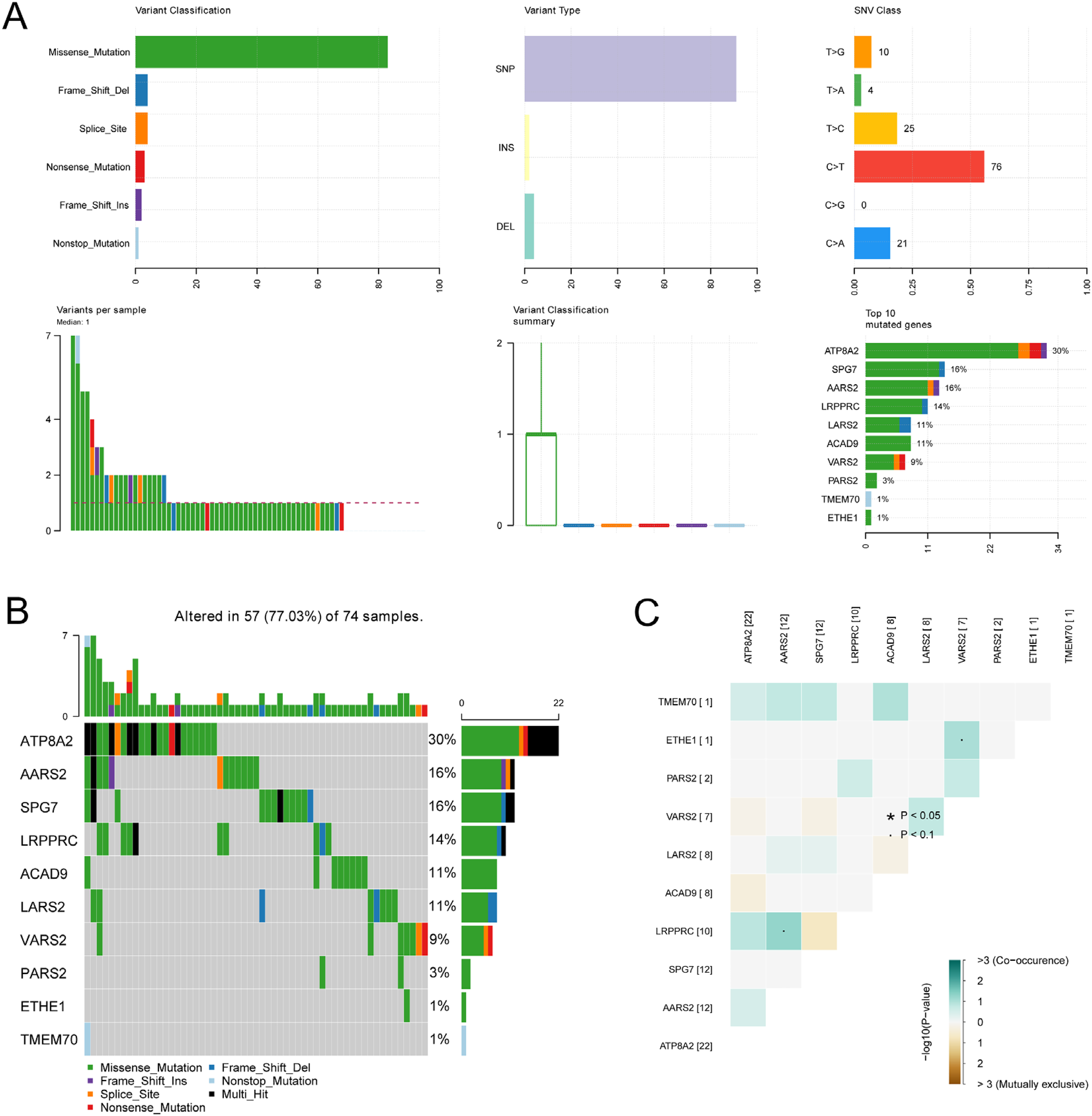
The mutation information of 10 differentially expressed-nuclear mitochondrial-related genes (DE-NMRGs). (A) Statistical presentation of various mutation types of methylation regulators. (B) Mutation waterfall diagram of DE-NMRGs in samples. (C) Co-mutation map of NMRGs.
Immune Cell Distribution and DE-NMRG Correlation
Immune infiltration in the COAD samples was calculated using CIBERSORT. In total, 22 kinds of immune cell types were obtained for analysis, and nine types of immune cells (B cell memory, B cell plasma, T cell CD8+, T cell CD4+ memory resting, T cell CD4+ memory activated, T cell regulatory (Tregs), NK cell activated, Macrophage M0, Myeloid dendritic cell activated) showed significant differences between the high and low risk groups (Figure 6A). Figure 6B shows the correlation between the characteristic NMRGs used to construct the RS model and the significantly different immune cells and scores. AARS2, ACAD9, LRPPRC, SPG7, and VARS2 were negatively correlated with stromal score, immune score and ESTIMATE score, while ATP8A2 was positively correlated with them. AARS2, ACAD9, LRPPRC, PARS2, SPG7 and VARS2 were positively correlated with tumor purity, while ATP8A2 was negatively correlated with tumor purity.

The distribution of various immune cells (A), and correlation analysis among immune cells, ESTIMATE score, and nine differentially expressed-nuclear mitochondrial-related genes (DE-NMRGs) (B) in two risk groups.
KEGG Pathways Related to Risk Grouping
Using the genome-wide expression levels of COAD samples from the TCGA dataset, KEGG pathways significantly associated with the risk groups were identified by GSEA. A total of 22 KEGG signaling pathways significantly associated with the risk group were obtained, including aminoacyl-tRNA biosynthesis (p = 0.004); butanoate metabolism (p = 0.018); β-alanine metabolism (p = 0.014); citrate cycle (TCA cycle) (p = 0.025); glycosaminoglycan biosynthesis chondroitin sulfate (p = 0.011); Notch signaling pathway (p = 0.009); peroxisome (p = 0.038); sucrose and starch metabolism (p = 0.038); leucine, valine, and isoleucine biosynthesis (p = 0.008); and leucine, valine, and isoleucine degradation (p < 0.001).
Validation of the Hub Genes With the HPA Database
As shown in Figure 7, expression levels of the nine hub genes (LARS2, PARS2, ETHE1, LRPPRC, TMEM70, AARS2, ACAD9, VARS2, and ATP8A2) were validated by immunostaining data from the HPA database. Compared with normal tissue, expression levels of PARS2, ETHE1, LRPPRC and ACAD9 increased in COAD tissue, whereas that of TMEM70, AARS2 and VARS2 decreased. Moreover, LARS2 was expressed at a medium level in both normal tissues and COAD tissues, while ATP8A2 was negative staining in normal tissues and COAD tissues. However, SPG7 was not found on the website.

Representative protein expression of the nine hub genes.
Mitochondria-Associated Gene Expression Proteins Alterations in Liver Metastasis Model
In order to verify the effectiveness of the mitochondrial genes screened in the above prognostic models for the development of colon cancer, a model of liver metastasis was constructed, in which Xeloda was shown to be an effective oral chemotherapy agent for advanced colon cancer. 34 Western Blot showed that the expression levels of LRPPRC and LARS2 were increased (p < 0.05; p < 0.001), the expression level of ETHE1 was not significant in drug group (Figure 7). However, the trend of change among the three groups was relatively obvious. These results suggested that mitochondria-related genes may be altered during the treatment of colon cancer and further proved the correlation between these genes and the prognosis of COAD (Figure 8).

The expression levels of mitochondria-associated proteins in liver metastasis model. (A) Western blot analysis of related targets, including LRPPRC (150 kDa), LARS2 (70 kDa), ETHE1 (25 kDa) and GAPDH (37 kDa). (B) changes in the protein expression of LRPPRC, LARS2 and ETHE1 were visualized by histograms. Relative protein expression was adjusted by GAPDH. Statistical evaluations were carried out by ANOVA. All data are represented as the mean ± SD of experimental values in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Mitochondria regulate factors related to tumorigenesis beyond energy production, such as oxidative stress and cell death regulation. 35 Many mitochondrial genes have been demonstrated as target genes contributing to cancers, such as MTHFD2 in diverse cancer cells, 36 SSBP1 in glioblastoma, 37 TFAM in colon cancer, 38 and mitochondrial NADH in non-small cell lung cancer and colon cancer. 39 However, the integrated role of NMRGS in predicting survival prognosis in COAD remains undefined.
In this study, a prognostic model was constructed. In the training datasets and the validation dataset of COAD, risk grouping was dramatically associated with prognosis after the samples were divided based on the RS model; moreover, the pathologic N and T stage and tumor recurrence were significantly different between the two groups. This indicated the validity of the RS model. The four prognostic factors of age, pathologic T stage, tumor recurrence, and prognosis in the nomogram model also showed excellent predictive performance. Then, an optimal combination of 9 DE-NMRGs was finally obtained, including LARS2, PARS2, ETHE1, LRPPRC, TMEM70, AARS2, ACAD9, VARS2, and ATP8A2. Recently, mitochondrial gene-related prediction models have been gradually reported. For example, Zhang et al identified three mitochondrial genes as predictive biomarkers for colorectal cancer. 16 Gao et al has made the first attempt to identify multi-gene markers of mitochondria-related genes and evaluate their potential function in COAD. 17 However, we constructed the prognostic model for COAD containing these nine genes at the first time. Our results, which showed OS has a greater AUC value at 1, 3 and 5 years, seem to have better prognostic efficacy.
LARS2, PARS2, AARS2, and VARS2 are involved in pathway of metabolism of proteins. LARS2 was found to regulate cell apoptosis and cell viability; participate in aminoacyl-tRNA ligase activity regulation, mitochondrial dysfunction, and accumulation of reactive oxygen species levels in granulosa cells; and play a role in tumor immunoevasion. 40 Abnormal expression of PARS2 contributes to tumor invasion. 41 AARS2 contributes to mitochondrial respiration and has been identified as a protective factor in the prognostic model of colorectal cancer. 42 Zhu et al suggested a gene signature containing AARS2 and VARS2 via involving mitochondrial respiration and proliferation in COAD. 43 VARS2 polymorphisms were found in a candidate prognostic marker for early breast cancer 44 and were associated with mitochondrial respiratory chain and oxidative phosphorylation. 45 Witherspoon et al indicated that overexpressed ETHE1 induces oxidative phosphorylation, mitochondrial respiration, and aerobic glycolysis in colorectal cancer. 46 Ozluk et al suggested that ETHE1 may promote the development of colon cancer and participate in sulfide catabolism and polysulfide formation by promoting the bioenergetics of tumor cells and the formation of polysulfide. 47 ATP8A2 and ETHE1 were also identified as prognostic biomarkers in lung adenocarcinoma. 48 Moreover, low expression of ATP8A2 is related to poor prognosis in lung adenocarcinoma, 49 and abnormally methylated ATP8A2 was found in various cancers. 50 The ATP8A2 level correlates negatively with serum levels of pro-inflammatory cytokine IFN-γ. 51 LRPPRC involves respiratory and ATP synthesis-related pathways. Highly expressed LRPPRC in colorectal cancer has been identified as a molecular biomarker, 52 and the accumulated LRPPRC promotes chemoresistance in colorectal cancer. 53 TMEM70 is involved in a metabolic shift via the Warburg effect and mitochondrial ATP synthase biogenesis, and could contribute to tumor promotion and progression. 54 As an inner mitochondrial membrane protein, TMEM70 is also important for mitochondrial organization. 55 The deficiency of ACAD9 induces mitochondrial dysfunction via energy metabolism and respiratory electron transport. 56 ACAD9 is regulated by LEF1 downstream of MYC and is involved in cell proliferation in colorectal cancer. 57 In addition, consistent with a more severe tumor microenvironment, the high-RS group has more inflamed immune features, such as T and NK cell activation. In summary, a survival prediction model containing LARS2, PARS2, ETHE1, LRPPRC, TMEM70, AARS2, ACAD9, VARS2, and ATP8A2 was successfully constructed, which may be used for forecasting the survival probabilities and clinical performance of COAD patients.
It is reported that the primary cause of death from colorectal cancer (CRC) is metastasis, with approximately 70% to 80% of CRC metastases occurring in the liver. 58 Our study findings indicated that the development of COAD was associated with mitochondrial metabolism and aminoacyl-tRNA biosynthesis, as determined through GO function and KEGG signaling pathway analysis. In the DE-NMRGs prognostic model, high expressed groups of ETHE1, LARS2, LRPPRC, PARS2, and TMEM70 exhibited a higher overall survival ratio. Immune-related analysis revealed that activated CD4+ memory T cells and regulatory T cells played a significant role in the progression of colorectal cancer, which correlated with LRPPRC, TMEM70, LARS2 and ETHE1. Therefore, a liver metastasis model was constructed and preliminarily validated the roles of LRPPRC, LARS2, and ETHE1 in the treatment of colorectal cancer. These three genes and their related pathways may demonstrate the central role in cellular metabolism, 59 energy production, 40 and maintaining intracellular environmental stability. 60 In mice treated with Xeloda, there was a significant upregulation of LRPPRC and LARS2 protein expression, suggesting these genes as potential efficacious targets for the drug action, which may contribute to prognosis and therapeutic efficacy evaluation for COAD patients. Studies have shown that low LRPPRC expression levels were associated with unfavorable outcomes in rectum adenocarcinoma, ovarian cancer, and kidney renal clear cell carcinoma, which had a substantial link with tumor immunity and pan-cancer immunotherapy. 61 LARS2 was verified to improve the prognosis of COAD by increasing CD4+ T infiltration. 62 The ETHE1 gene catalyzes the conversion of hydrogen sulfide into persulfides, thus safeguarding cells from the deleterious effects of toxic sulfide concentrations. 63 Although no significant correlation with Xeloda treatment was identified in this study of ETHE1, the trend is consistent with the model prediction and ETHE1 may still represent a crucial functional gene in other therapeutic regimen for COAD. 64 This underscores, from another perspective, the importance of examining the expression of the eight genes in the comprehensive prognostic model for colorectal adenocarcinoma. Further research is imperative to elucidate the mechanisms of these genes in the pathogenesis and treatment of COAD. This provides a research direction for identifying effective biomarkers for COAD patients clinically.
In current research, numerous biomarker models associated with cancer-associated fibroblast-derived genes, 65 metastasis-associated genes, 66 immune-related gene, 67 lipid metabolism-associated genes 68 have been utilized for predicting the diagnosis and treatment of CRC. Studies on the role of mitochondria-associated genes in predicting COAD prognosis are still insufficient. 17 Alterations in mitochondrial metabolic pathways may impact colon cancer cell proliferation and invasion. 43 We focused on researching mitochondria-associated genes and innovatively discovered nine biomarkers. The model demonstrated improved effectiveness in age, pathological T stage, tumor recurrence, and overall prognosis and received preliminary validation in a colorectal cancer liver metastasis model. Moreover, the genes in the predictive model are closely related to the development of immune cells in COAD, providing corresponding targets for the development of novel treatments such as immune checkpoint inhibitors. 69 However, our validation efforts are based primarily on data from public databases, which, while valuable, represent only a preliminary step in confirming our findings. The specificity and mechanisms of action of mitochondria-related genes in COAD require more in-depth molecular biology and clinical studies. These future studies are essential to solidify our understanding and to demonstrate the practical relevance of these genes in COAD treatment.
Conclusion
Our study has established a comprehensive prognostic model related to the mitochondria in COAD, marking the first report on the diagnostic and prognostic value of these nine genes. The 1-, 3-, and 5-year survival prediction AUC values in the Nomogram model also demonstrated superior performance. Functionally, the impact and significance of nuclear-mitochondrial-related genomic mutations on COAD were assessed, providing new multi-gene markers of COAD. Furthermore, this research offers essential insights and guidance for future investigations into the molecular underpinnings of COAD, crafting innovative treatment approaches, and improving the evaluation of patient prognoses.
Supplemental Material
sj-docx-1-tct-10.1177_15330338241258570 - Supplemental material for A Comprehensive Prognostic Model for Colon Adenocarcinoma Depending on Nuclear-Mitochondrial-Related Genes
Supplemental material, sj-docx-1-tct-10.1177_15330338241258570 for A Comprehensive Prognostic Model for Colon Adenocarcinoma Depending on Nuclear-Mitochondrial-Related Genes by Lingling Lv, Yuqing Huang, Qiong Li, Yuan Wu and Lan Zheng in Technology in Cancer Research & Treatment
Supplemental Material
sj-pdf-2-tct-10.1177_15330338241258570 - Supplemental material for A Comprehensive Prognostic Model for Colon Adenocarcinoma Depending on Nuclear-Mitochondrial-Related Genes
Supplemental material, sj-pdf-2-tct-10.1177_15330338241258570 for A Comprehensive Prognostic Model for Colon Adenocarcinoma Depending on Nuclear-Mitochondrial-Related Genes by Lingling Lv, Yuqing Huang, Qiong Li, Yuan Wu and Lan Zheng in Technology in Cancer Research & Treatment
Supplemental Material
sj-docx-3-tct-10.1177_15330338241258570 - Supplemental material for A Comprehensive Prognostic Model for Colon Adenocarcinoma Depending on Nuclear-Mitochondrial-Related Genes
Supplemental material, sj-docx-3-tct-10.1177_15330338241258570 for A Comprehensive Prognostic Model for Colon Adenocarcinoma Depending on Nuclear-Mitochondrial-Related Genes by Lingling Lv, Yuqing Huang, Qiong Li, Yuan Wu and Lan Zheng in Technology in Cancer Research & Treatment
Footnotes
List of Abbreviations
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Shanghai Ninth Peoples's Hospital, Shanghai Jiaotong University School of Medicine, No:SH9H-2021-A249-1.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: supported by the Construction of a Traditional Chinese Medicine Specialized Disease Alliance for Colorectal Cancer (ZY(2021-2023)-0302); Study on the Mechanism of Fuzheng Xiaoji Prescription Endogenously Activating TRG5 to Regulate Myeloid-Derived Suppressor Cells (MDSCs) and Remodel the Tumor Microenvironment in Colorectal Cancer Liver Metastasis; National Natural Science Foundation of China (No. 82274250 (2023-2026)); Fuzheng Xiaoji Progressive Formula Regulates TGF through VDR- β Research on the Mechanism of EMT Mediated Inhibition of Liver Metastasis in Colorectal Cancer; National Natural Science Foundation of China (No. 82104749 (2022-2024)); Study on the Molecular Mechanism by Which the Fuzhong Xiaoji Jinzhang Formula Downregulates METTL3-Mediated m6A Modification to Activate Ferroptosis and Inhibit the Growth and Liver Metastasis of Colorectal Cancer (No. 82305345 (2024-2026)).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.