
Abstract
Colorectal cancer (CRC) is one of the most prevalent malignant tumors of the digestive tract worldwide. Despite notable advancements in CRC treatment, there is an urgent requirement for preclinical model systems capable of accurately predicting drug efficacy in CRC patients, to identify more effective therapeutic options. In recent years, substantial strides have been made in the field of organoid technology, patient-derived organoid models can phenotypically replicate the original intra-tumor and inter-tumor heterogeneity of CRC, reflecting cellular interactions of the tumor microenvironment. Patient-derived organoid models have become an indispensable tool for investigating the pathogenesis of CRC and facilitating translational research. This review focuses on the application of organoid technology in CRC modeling, tumor microenvironment, and guiding clinical treatment, particularly in drug screening and personalized medicine. It also examines the existing challenges encountered in clinical organoid research and provides a prospective outlook on the future development directions of clinical organoid research, encompassing the standardization of organoid culture technology and the application of tissue engineering technology.
Keywords
Introduction
The pathology of colorectal cancer (CRC) has undergone significant advancements, leading to an expanded array of clinical treatment options for CRC patients, including endoscopic and surgical resection, radiotherapy, immunotherapy, palliative chemotherapy, targeted therapy, and extensive surgery and local ablation of metastases, which have been proven effective in suppressing CRC progression and prolonging the overall survival of CRC patients. 1 Implementation of population-based CRC screening, such as fecal occult blood testing and endoscopy, has also greatly improved overall survival rates and holds out the prospect of a higher cure rate for CRC.1,2 However, CRC remains the third most commonly diagnosed cancer and the second leading cause of cancer death worldwide in recent years due to the low level of actual screening prevalence, unpredictability of treatment effect, and the increasing incidence of CRC. 2 For instance, in 2020, CRC accounted for 10% of cancer cases and 9.4% of cancer deaths globally. Based on projections of aging, population growth, and human development, the number of new CRC cases is expected to reach 3.2 million globally in 2040. 1 The increased incidence of CRC is mainly attributed to the increased exposure to environmental risk factors due to the westernization shifts of lifestyle habits and dietary. 3 Therefore, the development of new therapeutic strategies through basic and clinical translational research is urgent.
The established cancer cell lines have been extensively used in research on tumorigenesis, tumor biology, and drug therapy of CRC. The application of the CRISPR-Cas9 gene editing system in cancer cell lines enables researchers to investigate the genetic mutations and pathogenesis of tumors in more depth. Despite the significant contribution of cancer cell lines to cancer research, they still have several limitations. On the one hand, each cell in the cancer cell lines is able to take full advantage of the nutrients in the culture medium as well as being exposed to antitumor drugs, which is not consistent with the tumor microenvironment in-vivo.4,5 Furthermore, cancer cell lines cannot fully recapitulate the physiological characteristics of tumors due to the absence of important components that constitute the tumor microenvironment, such as tumor mesenchymal cells, immune cells, fibroblasts, tumor vasculature, and extracellular matrix (ECM). 6 On the other hand, cancer cell lines would gradually lose the tumor genetic and intratumoral heterogeneity after multiple passages, which means that cancer cell lines cannot fully maintain the molecular and histological features of the original tumor tissue. 7 This partly explains why many anti-tumor drug screenings by established cancer cell lines eventually failed in subsequent animal experiments and drug clinical trials. 8 An alternative choice is to establish an animal model of the corresponding tumor, but the physiological environment of animals differs significantly from that of humans, a problem also faced by patient-derived tumor xenografts. Although patient-derived tumor xenografts indeed maintain partial morphological characteristics and heterogeneity of the corresponding tumor, high cost, time-consuming as well as low success rate may impede the experimental progress. 9
Intestinal organoids, a 3-dimensional (3D) cellular tissue that enables long-term growth and self-renewal in-vitro, are established by embedding isolated intestinal crypts or Lgr5-expressing intestinal stem cells (ISCs) into ECM and adding a special culture medium that contains essential growth factors.10,11 By varying the compositions of special culture medium, intestinal organoids are able to differentiate into almost all types of intestinal cells, comprising transient amplifying cells, Paneth cells, goblet cells, enteroendocrine cells, tuft cells, and enterocytes. 12 The biological phenotypes of intestinal organoids derived from adult stem cells (ASCs) in the intestinal crypts are highly consistent with those of the corresponding intestinal tissues, including gene composition and expression, protein expression pattern, biomechanical effects, and various biochemical characteristics.12,13 Organoids derived from pluripotent stem cells (PSCs) mimic in-vivo development by forming structures through processes that are exclusive to embryonic development. 14 Therefore, intestinal organoids are simplified and suitable 3D cell models in-vitro, which can be used to explore the pathology and treatment of various intestinal diseases including CRC. 15 In recent years, several teams have developed different culture methods to establish intestinal organoid models, and applied intestinal organoids to the study of gastrointestinal diseases, such as intestinal infection, inflammatory bowel disease, gastric cancer, and CRC.16–18 Intestinal organoids are widely used in in-vitro modeling, drug screening and testing, regenerative medicine, and personalized medicine.19–21
CRC organoids derived from tumor tissues or reprogrammed stem cells have become a vital tool for modeling tumor initiation, studying molecular mechanisms underlying tumorigenesis, and antitumor drug therapy. 22 Compared with cancer cell lines and animal models, CRC organoids retain tumor gene characteristics, heterogeneity, and intratumoral cellular heterogeneity, almost perfectly replicating CRC histological features. 23 Recently, exciting progress has been made in the clinical research of CRC organoids. Here, we review the application of organoid technology in CRC modeling, studying tumor microenvironment and guiding clinical treatment, especially in drug screening and personalized medicine, discuss the current challenges faced by clinical organoid research, and the future development directions of clinical organoid research are prospected from the aspects of the standardization of organoid culture technology and application of tissue engineering technology.
CRC Organoid Culture
Normal Intestinal Organoid Culture
ASC-Derived Intestinal Organoid Culture
Organoids can be established from 2 types of stem cells: ASCs derived from adult tissues; and PSCs, including induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs). The methods for establishing ASC-derived intestinal organoids and PSC-derived intestinal organoids are entirely different. Both ASC and PSC-derived intestinal organoids show strengths and weaknesses based on the scientific question and their range of preclinical/clinical applications. Therefore, researchers should take this into account before starting to study organoids.
The essential principle for culturing ASC-derived intestinal organoids is to mimic the microenvironment of adult ISCs, that is, the stem cell niche at the bottom of the crypt. The stem cell niche is the microenvironment necessary to maintain the self-renewal and proliferation of ISCs, which consists of various cells, ECM, and complex signaling pathways. An extremely rapid renewal rate is observed in the small intestinal epithelium, with a turnover time of 5 days. Actively proliferating Lgr5+ ISCs reside at the base of the crypt. 11 The crypts are mainly composed of ISCs, transit-amplifying cells, and Paneth cells. Within the crypts, the continuously dividing ISCs produce transit-amplifying cells, which can differentiate into absorptive intestinal epithelial cells and a variety of secretory cells, including Paneth cells (when transit-amplifying cells differentiate into Paneth cells, they can migrate downward back into the crypts and reside around ISCs), enteroendocrine cells, goblet cells, and tuft cells. 24
ISCs are strictly controlled by 4 signaling pathways. The Wnt signaling pathway plays a pivotal role in maintaining stem cell fate and driving the proliferation of both stem cells and transit-amplifying cells. Wnt ligand is produced by Paneth cells in the form of Wnt3. Wnt ligand has poor solubility, and the majority of signal transfer occurs by direct cell-to-cell contact, resulting in a gradient of decreasing Wnt activity from the bottom of the crypt to the villi. 25 Epidermal growth factor (EGF) signaling can strongly stimulate mitosis in both stem cells and transit-amplifying cells. Although EGF signaling regulates the division rate of ISCs, it does not seem to be necessary to maintain stem cell identity. 26 Notch contributes to maintaining the undifferentiated state of ISCs and transit-amplifying cells, Paneth cells provide essential Notch signal to ISCs by expressing Notch ligands DLL1 and DLL4. 27 Notch signal prevents the differentiation of ISCs into the secretory lineage, maintains ISCs stability as well as regulates the ratio between secretory and absorptive progenitors. 28 Bone morphogenetic proteins (BMPs) are members of the transforming growth factor-β (TGF-β) superfamily of ligands. The BMP signaling pathway inhibits stem cell proliferation and promotes cell differentiation. 29 In order to maintain the fragile balance between proliferation at the base of the crypt and differentiation above, the stem cell zone requires protection from the BMP signal. Therefore, BMP inhibitors, such as Noggin, Chordin-like 1, Gremlin 1, and Gremlin 2, are primarily secreted by myofibroblasts and smooth muscle cells located beneath the crypt, which establish a gradient of increasing BMP activity from the base of the crypt upwards.30,31 In short, the activation of Wnt and Notch signaling and the inhibition of BMP signaling are the necessary conditions to maintain the proliferation of ISCs, these signals are involved in regulating the abundance, lifespan, and activity of ISCs.
Based on the above findings, Sato et al established the first culture system enabling the growth of epithelial organoids from single Lgr5 + stem cells. 10 Samples from biopsies or resections from the small intestine and colon can be used to establish ASC-derived intestinal organoids. Two different approaches are available for dissociating crypts or single Lgr5 + stem cells from human intestinal tissue. One is to dissociate the crypts or Lgr5 + stem cells from the basement membrane using EDTA. 32 The other is to digest the intestinal tissue with collagenase to release the crypts or Lgr5 + stem cells. 33 Then, the isolated whole crypts or single Lgr5 + stem cells are embedded in Matrigel (provided the matrix component for attachment to form 3D organoids) and are cultured in a special medium based on Advanced DMEM/F12 medium and supplemented with Hepes, glutamine, N2, B27, N-acetylcysteine, antibiotics, R-spondin1, Noggin, and EGF (Figure 1).17,34 In mouse small intestinal organoid cultures, Wnt3 derived from Paneth cells is sufficient to maintain organoid growth if the Wnt potentiator R-spondin1 is added to the medium. For colon organoids culture, Wnt3a is essential due to the limited endogenous Wnt production by the colon epithelium. The introduction of Wnt3A to the combination of growth factors enabled limitless expansion of mouse colon organoids. However, the composition of the human small intestine and colon organoids culture medium is different from that of the mouse intestinal organoids culture medium. To achieve a long-term culture of human intestinal organoids, it was essential to supplement with gastrin and nicotinamide, as well as employ inhibitors of ALK and the p38 MAPK.17,35 A previous study found that gastrin and nicotinamide improved the organoid culture efficiency. 17 A83-01, the most commonly used Alk inhibitor, could inhibit the anti-proliferation effect of TGF-β and significantly improve planting efficiency. SB202190, the most commonly used p38 MAPK inhibitor, can block the negative feedback effect of p38 MAPK on the EGF receptor, thereby inhibiting goblet cell differentiation and increasing intestinal epithelial cell proliferation. Furthermore, the combination of these 2 compounds synergistically prolonged the culture period. Interestingly, human colon organoids were induced to differentiate along secretory and absorption lineages and generate various types of mature intestinal cells only when Wnt3a, nicotinamide, and p38 MAPK inhibitors were removed.17,34 Several groups have optimized the organoid culture conditions and improved the success rate and efficiency of organoid culture. For instance, Fujii et al found that using insulin growth factor-1 (IGF-1) and fibroblast growth factor-2 (FGF2) instead of p38 inhibitors can significantly improve the formation rate of human intestinal organoids and make them expand stably for more than 6 months. 35

Normal intestinal organoid culture and patient-derived CRC organoid culture. (A) The establishment of normal intestinal organoids. Intestinal crypts and intestinal stem cells are isolated by digestion of intestinal tissues using specific reagents. Intestinal organoids can be also derived from PSCs (ESCs and iPSCs) in a stepwise differentiation process. PSCs follow a guided differentiation towards an endodermal fate by exposure to Activin A, followed by the differentiation towards midgut/hindgut endoderm. Last, these organoids can be further directed towards the small intestine and colon depending on different growth factors. (B) The establishment of patient-derived CRC organoids. The top half of the image shows how to establish CRC organoids from tumor tissue. The bottom half of the figure shows how to establish CRC organoids from normal intestinal organoids or ESC/iPSC-derived organoids by engineering technology such as the CRISPR-Cas9 gene editing system.
Meanwhile, another research team developed an alternative method for culturing intestinal organoids. They employed an air-liquid interface (ALI) model, which entailed culturing neonatal mouse intestinal tissue fragments encompassing both epithelial and mesenchymal cells within an air-exposed collagen matrix. 36 These fragments were grown in a serum-containing medium but without specific growth factors. At present, the widely used ECM in organoid culture is Matrigel or basement membrane extract purified from Engelbreth-Holm-Sarm mouse sarcoma. The main components of Matrigel are laminin, type IV collagen, and nidogen, as well as a small amount of TGF-β, EGF, and IGF-1. Although Matrigel has been commercially produced and the price is acceptable, the composition of Matrigel is complex and there are differences among different batches of Matrigel, which will lead to poor repeatability of the experimental results. 37 Therefore, decellularized ECM, synthetic hydrogel, and engineered ECM proteins emerge at the right time, giving birth to organoid culture technology without Matrigel. 13 Table 1 summarizes the advantages and disadvantages of natural and synthetic ECM used in intestinal organoid culture. In 2019, Giobbe et al 38 designed ECM hydrogels derived from decellularized porcine small intestine mucosa/submucosa, they found that human small intestine stem cells could grow and eventually generate human small intestinal organoids in these ECM-derived hydrogels. Biological hydrogels synthesized based on materials such as hyaluronic acid and gelatin can also replace Matrigel for the culture of CRC organoids and be applied in the co-culture system. 39 By quantitative regulation of matrix stiffness, matrix stress relaxation rate and matrix integrin-ligand concentration, Heilshorn et al used engineering techniques to synthesize an engineered ECM-hyaluronic acid elastin-like protein, in which intestinal epithelial organoids can grow, differentiate, and passage. 40 This engineered ECM has good repeatability and supports biodegradation, which provides a promising alternative ECM for current organoid research. The development of these synthetic ECMs brings hope for the possibility of clinical use of human intestinal organoids.
Natural and Synthetic Extracellular Matrix Used in Intestinal Organoid Culture.

PSC-Derived Intestinal Organoid Culture
In principle, to generate an organoid, it is necessary to faithfully replicate the entire process of organ development from PSC. However, it is practically challenging to provide all necessary biochemical cues for cell differentiation and tissue assembly in-vitro. Thankfully, cells in-vitro tend to follow a semi-autonomous differentiation trajectory similar to in-vivo. Creating PSC-derived organoids requires a stepwise differentiation process, in which growth factors are sequentially introduced to mimic the cellular signaling observed during embryo development. These organoid cultures initiate by inducing ectoderm, mesoderm, or endoderm tissues and subsequently recapitulate the processes observed during early embryonic development, extending through the later stages of fetal development. 41 Some PSC-derived organoids may contain cells from multiple germ layers to closely mimic the corresponding organ in-vivo. In general, the formation of PSC-derived organoids involves 3 essential steps. Firstly, specific signaling pathways responsible for developmental patterning are activated or suppressed using morphogens and signaling inhibitors. This is done to establish accurate regional identities during stem cell differentiation. Subsequently, media with specific growth and signaling factors are devised to support the proper terminal differentiation of desired cell types within the organoid. Finally, cultures are cultivated in a manner that enables their growth in 3 dimensions, achieved either through cellular aggregation into 3D structures or embedding the cultures in a 3D matrix. 42 Specifically, PSC-derived intestinal organoids were established by initially treating human PSCs with Activin A to drive endodermal identity, followed by incubation with FGF4 and Wnt3a to promote hindgut specification, eventually addition of EGF and Noggin to support the proper terminal differentiation (Figure 1).43,44 The duration of this differentiation takes approximately 2 months. These organoids were composed of proliferative zones resembling crypts, which expressed ISC markers and exhibited the ability to differentiate into all cell types of the intestinal epithelium. 44 A major advantage of this culture system is that PSC has the potential to differentiate into all lineages within the tissue. The clinical utility of organoids derived from PSC is restricted due to the genomic instability resulting from exposure to reprogramming factors, as well as their tendency to exhibit a relatively premature phenotype. Therefore, organoids derived from PSC typically serve as valuable models for studying tissue development.
Explorations on the Improvement of Intestinal Organoid Culture
In response to the need for research on the interaction of the intestinal epithelium with environmental factors, several attempts have been made to modify the shape and structure of the intestinal organoids. Intestinal organoids cultured in Matrigel exhibit an apical surface (lumen) facing inward enclosed within the organoids with a basolateral surface facing the outside of the organoid. Thus, the challenge for researchers using intestinal organoids to study the interaction of epithelium with environmental factors such as nutrients, microbes, and bacterial metabolites is that the apical/luminal surface of the epithelium is enclosed within the organoids and difficult to access. Moreover, this is not conducive to investigating invasion sites and cell tropism of pathogenic microorganisms as well as performing drug toxicity assays. 45 Microinjection of nutrients, microbes, or bacterial metabolites into the lumen of the organoids may be a viable method to solve this problem, as some studies performed (Figure 2(A)).46,50 This can be a time-consuming, labor-intensive, and challenging method and may result in spillage of microinjected contents into the basolateral space. In addition, it is difficult to realize the standardization of exposure to microinjected substances as the irregular shape and size of organoids and the potential interactions with accumulated contents of the lumen. Interestingly, Williamson et al designed an automated high-throughput computer-driven microinjection platform for reagents of interest delivery to the lumen of organoids and high-content sampling. 51 But this system still cannot be widely available because it requires complex and expensive equipment. Another method to access the apical surface of intestinal organoids is to generate a 3D-derived polarized monolayer on a Transwell permeable support (Figure 2(B)).47,48 Long-term self-organizing 2-dimensional (2D) epithelial monolayers with highly differentiated epithelial cells can be established by exposure of the apical surface of a 2D monolayer to an ALI (Figure 2(C)). 48 This 2D culture method allows independent access to the apical and basolateral surfaces, but it requires large numbers of cells and is difficult to visualize with a microscope without disassembling the Transwell system. To address these issues, Co et al developed a method to reverse intestinal organoid polarity by removing ECM scaffold proteins and suspension culturing in low-attachment plates, thus enabling access to the apical epithelium (Figure 2(D)). 49 This resulting apical-out organoids could maintain integrity and barrier function as well as appropriate polarity, and exhibit polarized absorption of nutrients and other substances. 52 Potential applications of this apical-out organoid model include evaluating the epithelial barrier integrity, exploring nutrient uptake, and studying microbe-epithelium interactions.
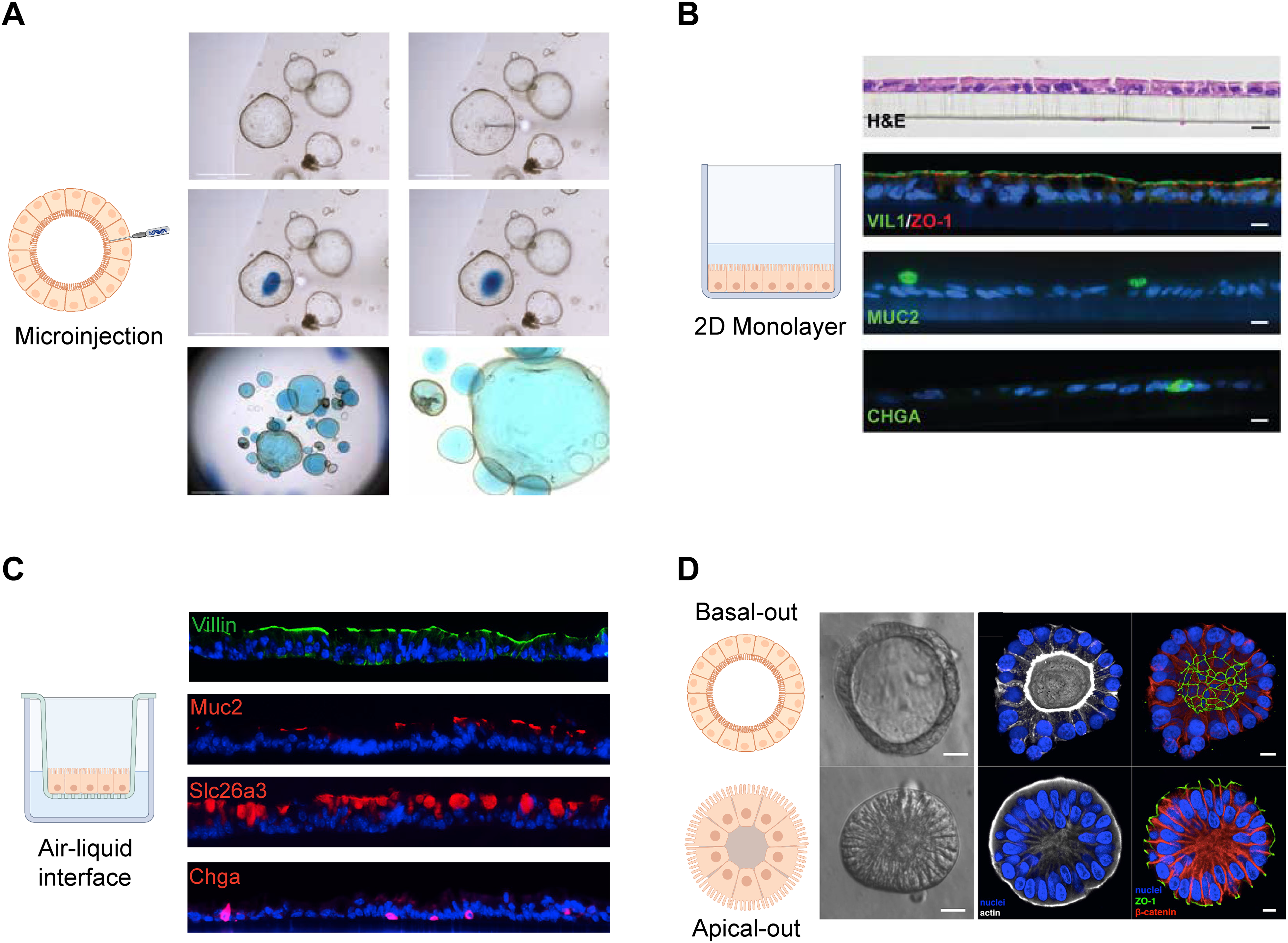
The explorations on the improvement of intestinal organoid culture. (A) The process of injecting an organoid with FastGreen dye by Puschhof et al and overview images of a dome with injected organoids. 46 Scale bars, 2.5 and 0.5 mm, respectively. (B) Organoids were dissociated and seeded onto coated Transwell membranes to form epithelial monolayers by VanDussen et al. 47 Paraffin sections from rectal epithelial organoids monolayers were stained for H&E or immunostained for VIL1 and ZO-1, MUC2 or chromogranin A (CHGA) to visualize differentiated cell types. Scale bars, 10 μm. (C) In-vitro culture of a self-organizing colonic epithelial monolayer. 48 Colon organoids were dissociated into single cells and plated onto Transwell membranes. Then the cells were exposed to air-liquid interface (ALI) for 21 days after being submerged in conditioned medium for 7 days. Sections of ALI d21 monolayers stained for Villin (brush border), Slc26a3 (colonocytes), Muc2 (goblet cells), and Chga (enteroendocrine cells). Scale bars, 50 μm. (D) Organoids in suspension culture established by Co et al exhibit Apical-out polarity. 49 Basal-out organoids and Apical-out organoids are depicted schematically. Organoids were imaged using modulation contrast microscopy and using confocal microscopy. Nuclei in blue, actin in white, ZO-1 in green, and b-catenin in red. Scale bars, 10 μm.
CRC Organoid Culture
Human ASC-derived CRC organoids can be generated not only from surgically removed tumor specimens but also from routine biopsy specimens. 23 The method of establishing organoids from human CRC tissues is similar to that of normal intestinal organoids (Figure 1). In short, cancer stem cells were first isolated from CRC tumor tissue, then embedded into the Matrigel, and CRC organoid medium was added for several passages and subsequent experimental analysis. 53 The medium composition of human normal colon organoids can be used to culture human CRC organoids after subtle adjustment.17,54 Wnt3a and R-spondin1 are not essential for maintaining the long-term expansion of human CRC organoids, because the Wnt/β-catenin signaling pathway in most CRC has mutations in key genes that cause abnormal activation of the tumor-derived organoids in CRC patients. Due to the accumulation of mutations during adenoma-tumor progression, the dependence of tumor cells on niche factors is reduced, and the culture of patient-derived tumor organoids does not depend on some specific growth factors in the medium.55,56 Interestingly, deAngelis et al 57 successfully cultured organoids from circulating tumor cells, and the organoids derived from circulating tumor cells can be used as models to study the characteristics of metastatic CRC and discover new biomarkers. In addition to the above conventional culture methods, Li et al 58 innovatively used the ALI method for CRC organoid modeling. The advantages of the ALI culture method include improved in-vitro oxygenation and the ability to perform organ physiological studies under controlled conditions. The CRC organoids derived from ASC possess a simple structure and can be established efficiently within a relatively short time. Moreover, they can recapitulate the cancer genome changes of specific patients. These qualities contribute to their widespread usage as powerful tools and make them highly suitable candidates for various clinical applications.
CRC organoids can also be generated from PSCs. To date, many PSC-derived organoids have been successfully created. However, there is a vital limitation when using PSC-derived organoids for cancer research. These organoids do not possess the acquired cancer gene mutations that ASC organoids derived directly from tumor tissue and preserved in culture. To solve this problem, some research teams attempt to directly reprogram cancer cells into PSCs to understand neoplastic processes. However, there are relatively few successful cases. Alternatively, another strategy involves engineering oncogenic mutations into PSCs via CRISPR-Cas9 editing, which are then induced to differentiate into specific organs. This approach has been used primarily to mimic cancers originating in the brain, retina, lung, and pancreas.59–62 Last, normal somatic cells from patients with cancer predisposition syndrome were reprogrammed into PSCs for use in mimicking cancers from patients with specific genetic mutations. Familial adenomatous polyposis (FAP) is an inherited form of CRC, caused by mutation of the adenomatous polyposis coli (APC) gene. Recently, Crespo et al created iPSCs from the skin fibroblasts of 2 patients with FAP and then used colon organoids derived from FAP-iPSCs to study the role of genetic factors in the progression of CRC. 63 The results showed that the colon organoids derived from FAP-iPSCs exhibit elevated Wnt signaling activity and heightened levels of epithelial cell proliferation compared to APC-wild-type organoids, which are indicative of FAP-associated polyp formation and the progression toward CRC. The resulting colon organoids can be used as a platform for drug discovery and testing. Meanwhile, Sommer et al utilized human iPSCs from FAP-specific APC mutant individuals to generate intestinal organoids as well and reported similar findings of APC mutations impacting differentiation in intestinal organoids. 64
Applications of Organoids in CRC
With the development of organoid technology, organoids have become a useful model system for CRC disease model construction, pathogenesis research, tumor microenvironment research, drug screening, drug toxicity testing, and individualized therapy. Figure 3 summarizes the application of organoid models in CRC studies.

The applications of organoids in CRC. (A) CRC modeling. CRISPR-Cas9 gene editing system can be used to accurately edit specific genes or genomes of normal intestinal organoids to establish engineering organoids. After xenotransplantation of the resulting organoids into mice, they can simulate the tumor-generating process in-vivo. (B) Establishment of living biobank. A CRC organoid living biobank can be established by collecting intestinal tissue specimens from many CRC patients and culturing the corresponding CRC organoids. Living biobanks have great potential for applications in new drug development and drug screening. (C) Deciphering tumor microenvironment by co-culture with immune cells, fibroblasts, and microorganisms. With the help of microfluidic technology and organoid chips, high-throughput drug screening and testing as well as personalized medicine can be achieved. The figure was created with Biorender.com.
CRC Modeling
Organoids show good genetic and epigenetic stability.65,66 Organoids derived from primary and metastatic CRC all retain the genetic diversity and morphological stability of their parent tumors.23,67 CRC is generated by mutations in genes related to signaling pathways such as Wnt/β-catenin, TGF-β, TP53, RAS-MAPK, and PI3K.68,69 CRISPR-Cas9 gene editing system is a breakthrough technology, which is used to achieve accurate editing of specific genes or genomes of normal intestinal organoids and simulate the tumor-generating process in-vivo. CRC occurs through the adenoma-adenocarcinoma pathway and serrated pathway. The adenoma-adenocarcinoma pathway is a classic pathway for CRC occurrence. According to this model hypothesis, the mutation of the APC gene activates the Wnt signal, leading to the development of colon polyps in the intestine, and subsequent mutations of SMAD4, TP53, and other genes induce colon polyps to transform into invasive and metastatic phenotypes. 70 The establishment of colonic organoids carrying various gene mutations via CRISPR-Cas9 technology can be applied to observe the effects of gene mutations. Drost et al 55 used CRISPR-Cas9 technology to target and modify oncogenes Kras and tumor suppressor genes APC, P53, and SMAD4 and then screened mutated intestinal organoids by removing specific niche factors from the culture medium. Only organoids carrying 4 gene mutations can survive and grow up in media lacking all niche factors such as Wnt3a, Noggin, R-spondin1, and EGF. After in-situ transplantation of these mutant organoids into immunodeficient mice, the organoids with APC, Kras, and TP53 mutations but without SMAD4 mutations only formed adenomas, while the organoids with 4 gene mutations successfully formed adenocarcinoma after transplantation. This study preliminarily established a CRC organoid model of the adenoma-adenocarcinoma pathway. Similarly, Matano et al 71 also created a genetically modified organoid by using CRISPR-Cas9 technology. Except for modifying the 4 mutated genes above, the PIK3CA mutation was further introduced, then the gene-edited intestinal organoids were implanted into mice, and eventually tumors were successfully developed. These studies demonstrate that colonic organoids carrying successive mutations in the APC and TP53 genes exhibit aneuploid characteristics, suggesting the progression of CRC in chromosomal instability pathways. Persistent chromosomal instability is closely related to the evolution of CRC organoid tumors. 72
The established method of the CRC organoid model of the serrated pathway is similar to that of the classical approach. The serrated pathway is initiated by a mutation in the BRAF oncogene. 73 Fessler et al 74 established a model of serrated adenoma by introducing BRAFV600E mutations into intestinal organoids. Kawasaki's team 75 further used CRISPR-Cas9 technology to transfer the chromosome rearrangement containing the R-spondin gene into human normal colonic organoids, and at the same time transferred into BRAFV600E mutation, the resulting organoids formed flat sawtooth lesions. Except for the fusion protein of BRAFV600E and the R-spondin gene, the organoids overexpressing the Grem1 gene formed tumors with the characteristics of serrated adenoma after implantation in mice. In 2019, Lannagan et al 76 developed an organoid model with multiple gene mutations with the help of CRISPR-Cas9 technology to simulate the serrated pathway. By analyzing the gene alteration characteristics of serrated CRC in the TCGA gene database, Tgfbr2, ZnRF2, RNF43, CdKN2a, and BRAFV600E were selected as the target genes for CRISPR-Cas9 editing. These modified genes were introduced into mouse colonic organoids, and then these organoids were transplanted into the colon of immunodeficient mice. Nearly half of the mice transplanted with BRAFV600E and Tgfbr2 mutant organoids grew adenocarcinoma, and these mice survived more than 3 months. Almost all of the mice transplanted with 5 target gene mutant organoids developed adenocarcinoma, and their survival time was shortened to 4 weeks. In order to explore the origin of CRC-related mutation characteristics, Drost et al 77 used CRISPR-Cas9 technology to knock out a key DNA repair gene MLH1 in human colonic organoids. The whole gene sequencing results showed that the mutation characteristics in MLH1-deficient colonic organoids were similar to those in CRC with mismatch repair defects, suggesting that they successfully established a microsatellite unstable CRC model.
It is essential to transplant genetically edited engineered organoids into immunodeficient mice in order to explore and clarify the contribution of specific gene mutations to CRC metastasis. A 2018 study found that compared with other mutant gene combinations, transplantation of intestinal organoids with APC, Kras, and Tgfbr2 mutations into the spleen of immunodeficient mice had the highest incidence of liver metastasis. 78 This suggests that the activation of Wnt and Kras in intestinal epithelial cells and inhibition of TGF-β signal are sufficient to induce liver metastasis of CRC. Several research teams used engineered organoid transplantation to establish a complex model of metastasis from colon adenoma to colon adenocarcinoma and then to distant metastasis, which completely summarized the progress of CRC.79,80 The work of these teams has contributed valuable experience to genetic research and preclinical research. Moreover, Roper et al 81 confirmed that both genetically engineered colonic organoids and patient-derived CRC organoids can form liver metastasis after orthotopic transplantation to the distal colon. In short, gene-edited colonic organoids are excellent tools for studying the pathogenesis of CRC and exploring the role of gene mutations in diverse signal pathways. These patient-derived CRC organoid models have great application prospects in exploring disease progression, screening antineoplastic drugs, and formulating individualized and precise treatment strategies.
Decipher Tumor Microenvironment
The establishment of the CRC organoid model has enabled the study of tumor microenvironment to be undertaken in depth. The immune cells, stromal cells, cancer-associated fibroblasts, tumor vasculature, commensal microbiota, and ECM are significant components of the tumor microenvironment. 82 Importantly, the components of the tumor microenvironment are able to maintain the stemness of cancer stem cells and affect the occurrence, progression, metastasis, and drug resistance of CRC. 83 Tumors have the ability to change the surrounding environment, which is an essential basis and unique feature of distant metastasis. To date, most CRC organoid models only have tumor cells and lack an actual tumor microenvironment to mimic in-vivo environments. Therefore, co-culture systems have been built to explore the interaction between tumor microenvironment and CRC organoids.
As one of the most critical components of the tumor microenvironment, immune cells are closely related to the occurrence, development, and treatment of CRC. 84 There are 2 methods for tumor organoids to mimic tumor microenvironment. One is to establish tumor organoids to simulate tumor microenvironment through the method of gas-liquid interface. For example, Neal et al 85 used the ALI method to integrate primary tumors and tumor-infiltrating lymphocytes to simulate the toxicity of antibodies against programmed death receptor 1 and programmed death factor ligand 1 to organoids. The other method is to mimic the tumor microenvironment with the help of a microfluidic chip which can perform high-throughput analysis. Zheng et al 86 designed a microfluidic chip, that can accurately regulate the oxygen concentration in each chamber on the chip, successfully simulate the anoxic environment of lung cancer organoids and the aerobic environment of liver organoids in-vitro, and achieve the hypoxia-induced tumor metastasis, which provides a good model for the screening of antineoplastic drugs in anoxic environment. Cancer immunotherapy has been a potential tumor therapy in recent years. Dijkstra et al 87 established a protocol for the isolation of tumor-reactive T cells. Tumor-reactive T lymphocytes were obtained by co-culturing CRC organoids prestimulated by interferon γ and lymphocytes in peripheral blood. When transplanted into the body, these T cells enable to help eliminate tumors in the patient's body. This protocol may be used to estimate the in-vitro efficacy of tumor immunotherapy. The recent study by Schnalzger et al 88 developed a platform to test the cytotoxicity of chimeric antigen receptor cells to colon organoids derived from patients. Epithelial cell adhesion molecules, which are commonly expressed by chimeric antigen receptors targeting epithelial cells, were introduced into NK92 cells, and then co-cultured CRC organoids expressing EGF receptor variant III (a new tumor antigen) and chimeric antigen receptor NK92 cells targeting EGF receptor variant III. The results showed that chimeric antigen receptor-NK92 cells could specifically kill CRC organoids without damaging normal organoids. Therefore, the above studies show that co-culture of CRC organoids with immune cells can not only study the interaction between immune cells and tumor but also test the efficacy of tumor immunotherapy to develop novel immunotherapy strategies.
Cancer-associated fibroblasts (CAFs) are mainly derived from fibroblasts in ECM. Fibroblasts can initially block the development of early CRC, but they are transformed into CAFs under the action of TGF-β in the tumor microenvironment. 89 It has been reported that under hypoxia, extracellular vesicles derived from fibroblasts may induce colony formation of CRC organoid cells by transmitting Wnt and EGF signals, and then promote the occurrence of CRC.90,91 In 2017, Öhlund et al 92 co-cultured human organoids derived from the tissue of pancreatic ductal adenocarcinoma with mouse CAFs and the results showed that CAFs formed a different subgroup. They are situated at the distal end of the tumor and can secrete inflammatory factors such as interleukin-6, thus revealing the heterogeneity of CAFs. Therefore, CAF subsets that promote tumorigenesis should be selectively targeted in the treatment of CRC.
To sum up, the co-culture of CRC organoids with immune cells and fibroblasts has a certain value and application prospect in tumor microenvironment research and development of tumor immunotherapy strategies. Organoid co-culture with other cell types can improve the biological complexity of these systems, which is one of the future research directions. And, these complex models will be vital for the establishment of preclinical drug evaluation platforms used in Phase I and Phase II clinical trials.
Drug Screening
One of the most significant roles of organoid research is to provide evidence and guidance for clinical drug treatment decisions. The CRC organoid model better conserves the intratumoral heterogeneity and genetic heterogeneity of the original CRC tissue and brings more credible experimental results. Recently, some excellent studies have confirmed the great potential of CRC organoids in guiding clinical drug therapy.
Organoids provide a new platform for in-vitro drug screening and drug efficacy testing. vandeWetering et al 93 screened CRC organoid drugs by establishing an organoid biobank, which included organoids derived from tumor tissues of 20 CRC patients. In this study, 83 therapeutic drugs, including chemotherapy drugs, targeted drugs, and clinical trial drugs, were utilized to treat CRC organoids. The results showed that the biobank platform of this kind of organoids could complete high-throughput drug screening and identify the correlation between drugs and different genes. For instance, CRC organoids with RNF43 mutations are sensitive to Porcupine inhibitors, while CRC organoids with Kras mutations are resistant to tyrosine kinase receptor inhibitors. This study establishes the potential of CRC organoids for drug screening applications. Kondo et al 94 used organoids derived from CRC patients to complete screening tests on 2427 drugs and they found that CRC organoids showed different sensitivities to different drugs, just as various drugs have inconsistent therapeutic effects in humans, indicating that CRC organoids can faithfully predict drug efficacy in-vivo. Du et al 95 designed a higher throughput organoid drug screening platform and efficiently completed screening tests for thousands of drugs, accelerating organoid research and drug discovery. These studies demonstrate the potential of organoids for high-throughput drug screening analyses to develop new treatment options.
For oxaliplatin-resistant CRC patients, the selection of subsequent molecular targeted drugs and the prediction of efficacy are major challenges for clinicians. To address this problem, Shen et al 96 used a patient-derived CRC organoid model to conduct in-vitro drug screening experiments, and they successfully screened out a Kruppel factor 5 inhibitor ML264, which retained the sensitivity of drug-resistant CRC organoids to oxaliplatin by restoring cell apoptosis in CRC organoids. This is another convincing demonstration of the role of CRC organoids in screening for potential therapeutic drugs. In addition, organoids derived from human induced pluripotent stem cells can also be used as CRC models for drug testing. 63 Organoids, as preclinical models, accelerate the process of new drug research and development and reduce the cost of new drug research and development. CRC organoid model can test the effectiveness of drugs undergoing clinical trials, and as a supplement to clinical trials, promote the process of new drug research and development. 97
Drug Toxicity Testing
Drug toxicity is the main reason for the failure of drug development. Traditional human cell lines have been the most common model used to study drug efficacy, mechanisms, and toxicity. Drug-induced gastrointestinal toxicity is one of the most commonly occurring adverse effects in clinical studies. 98 However, the cell line and animal model cannot accurately predict the adverse effects on the human body. Intestinal organoids have become a promising candidate tool to develop new drugs and evaluate drug toxicity. 99 In general, the toxicity of drugs (especially antitumor drugs) includes toxicity to normal intestinal tissues (surrounding tumors) and toxicity to vital organs of the body such as the liver, kidney, heart, and brain.
To assess the effect and toxicity of KAN0438757, a newly developed PFKFB3 inhibitor targeting glycolysis, Oliveira et al 100 treated patient-derived CRC organoids and patient-derived normal colon organoids with the compound. Then they observed changes in organoid morphology and growth using microscopy to evaluate its impact. The results revealed that normal colon organoids maintained their morphology, while CRC organoids demonstrated a significant impact on their structure, displaying disintegration, a significant reduction in size, and an increased count of surrounding single cells. These alterations suggest the occurrence of induced cell death and reduced organoid viability. However, determining cell viability by morphology is qualitative and non-objective. Methods and indicators to quantify organoid cell viability are required. For instance, Park et al examined the toxicity of SiO2 and TiO2 on colon organoids by determining the cell viability of the organoids by CellTiter-Glo 3D cell viability assay and quantifying apoptosis by WB detection of Bax/Bcl-2 protein ratio. 101 Engineered nanoparticles are widely used in various products, intestinal organoids allow quick screening of the engineered nanoparticles that can be safely ingested into the body. Markus and his colleagues recently investigated the toxicity of some nanoparticles on intestinal organoids by monitoring barrier integrity via measuring transepithelial electrical resistance, assessing cell viability via MTT assay, and detecting oxidative stress as well as inflammatory response (IL-8 level). 102 While immunotherapy using chimeric antigen receptor (CAR)-engineered lymphocytes has demonstrated remarkable outcomes in treating leukemia, new preclinical models are required to test CAR-mediated cytotoxicity in a tissue-like environment for solid tumors like CRC. A recent study developed a method to quantitatively evaluate the CAR-mediated cytotoxic activity against 3D organoids without single cell digestion. Luciferase/GFP expression was introduced into organoids by using lentiviral transduction and the luciferase activity was measured using specific equipment. The remaining luciferase inside the cells could accurately reflect the overall cell viability. 88
The unpredictable liver toxicity of certain drugs can result in liver failure among patients, which is a significant contributing factor to the failure of drugs in clinical trials. Drug-induced liver injury is a frequent cause of liver failure globally, which can be categorized into intrinsic and idiosyncratic liver toxicities.103,104 Direct intrinsic liver toxicity typically depends on the dosage, whereas idiosyncratic liver toxicity is unpredictable and can often occur within therapeutic doses. Liver organoid models have been established to predict these 2 types of drug-induced liver injury. The assessment methods of hepatic toxicity of drugs include measuring hepatic enzyme activity, hepatocyte structures, cell viability, and redox status. The cytochrome P450 enzyme family is a crucial drug metabolizing enzyme in the human liver and metabolizes the majority of drugs within the liver. Metabolites produced by cytochrome P450 enzymes could be hepatotoxic, potentially affecting various components in hepatocytes, leading to cell stress and alterations in cellular structure or function. Changes to nuclei and mitochondria can cause mitochondrial respiratory dysfunction, resulting in the production of reactive oxygen species, which can then damage liver cells and cause detrimental effects on liver health. Mekky et al employed human liver organoids to examine the toxicity of magnesium oxide nanoparticles coated with aspartic acid and valproate. The findings revealed that both drugs have toxic effects on the liver organoids by reducing cell viability, lowering ATP levels, and elevating reactive oxygen species. 105 Liver organoids derived from human iPSCs have been generated in a 3D perfusable micropillar chip. 106 The authors observed that the activity of cytochrome P450 enzymes in these organoids was higher than that of organoids grown in static conditions, indicating that the perfused culture system facilitated the hepatic enzymatic activity. The researchers then evaluated the cell viability of liver organoids treated with acetaminophen using a CCK-8 assay. The results showed that high-dose acetaminophen treatment markedly decreased the cell viability of the liver organoids. Compared to traditional organoid cultures, organoid-on-a-chip offers the advantage of enabling customized access to each organoid and allows for efficient toxicity testing through varying concentrations, various dosages, and repetitions while remaining cost-effective and straightforward.
To test whether human ESC-derived kidney organoids could be used to study drug toxicity, Morizane et al 107 treated the organoids with gentamicin and cisplatin, which have certain kidney toxicity. The expression of Kidney Injury Molecule-1, LTL, and E-cadherin, biomarkers for proximal and distal tubule injury, respectively, was then detected in the organoids by immunostaining. The results showed that gentamicin injured proximal tubules in the organoids and cisplatin exhibited proximal and distal tubular toxicity. Kidney organoids derived from iPSC established by Takasato et al successfully mimicked acute apoptosis similar to in-vivo after treatment with cisplatin compared to untreated control organoids. 108 Apoptotic cells were detected by cleaved caspase 3 antibody-staining in this study, and the severity of apoptosis was quantified by the number of apoptotic proximal tubules. Furthermore, engineered heart organoids with improving maturity could be used to detect potential drug toxicity in the heart. 109 For instance, Richards et al demonstrated that human cardiac organoids enable the recapitulation of the responses that hypoxic cardiac injury aggravated the cardiotoxicity of doxorubicin. 110 Despite the advantages, organoids are not extensive models and do not consider organ-organ interaction. Moreover, organoids have intra- and inter-batch phenotype variability. 111 Overall, when the next generation of cancer organoid cultures is standardized, culture reproducibility is improved, and organoids can accurately mimic tumor heterogeneity, organoid models will play a critical role in drug toxicity testing.
Personalized Medicine
Personalized tumor therapy is an important step toward achieving precision medicine. The benefits of individualized therapy include reducing unnecessary treatment attempts, reducing preventable adverse effects, and preventing the emergence of drug resistance. The time required to establish a CRC organoid model is shortening, with drug test results available within weeks. After the causes of drug resistance in CRC have been analyzed, truly effective treatment strategies could be elaborated. The presence of potential drug targets and signal pathway abnormalities in CRC organoids is a prerequisite for individualized therapy. Another vital condition is to establish whether the results of in-vitro drug sensitivity or resistance tests based on CRC organoids match the real situation in CRC patients. 112 Therefore, the drug response in the CRC organoid model requires to be compared with the actual patient response. Weeber et al 23 performed whole-genome sequencing on organoids derived from patients with metastatic CRC and the results showed that 90% somatic mutations were identical between CRC organoids and biopsy specimens, with a DNA copy number correlation coefficient of 0.89. This finding demonstrates that CRC organoids basically retain the genetic and phenotypic characteristics of the original tumors and that the application of organoids to individualized tumor therapy is feasible. Organoid living biobanks offer the opportunity to test drug safety and efficacy. In 2018, Vlachogiannis et al 19 used organoids derived from 71 metastatic CRC patients to establish a living biobank. The results of the analysis of the organoids phenotype and genotype indicated that the patient-derived organoids were highly similar to the original tumors, and the response of CRC organoids to drugs was largely consistent with that of patients to the same drugs, suggesting the predictive value of organoids in individualized medicine. In 2019, Ooft et al 113 tested the ability of CRC organoids to predict the efficacy of chemotherapy drugs. CRC organoids could predict the efficacy of 80% patients treated with irinotecan chemotherapy but failed to accurately predict the efficacy of patients treated with 5-fluorouracil and oxaliplatin. This indicates that organoids have different predictive abilities for the efficacy of different chemotherapy drugs. Recently, the study of Yao et al 114 showed that organoids enable to prediction of the chemoradiotherapy response of patients with locally advanced rectal cancer. The accuracy, sensitivity, and specificity of CRC organoids in predicting the efficacy of chemoradiotherapy were 84.43%, 78.01%, and 91.97%, respectively. However, it should be pointed out that the organoids derived from 19 patients in this study responded well to radiotherapy and chemotherapy, but the tumor tissue of these patients was resistant to radiotherapy. This may be attributed to the intratumoral heterogeneity of the tumor or tumor microenvironment. Schumacher et al 115 evaluated the effect of tumor heterogeneity on the activity of Kras/MAPK signaling pathways in CRC organoids and on the response to EGFR-targeted drugs. Targeted proteomics analysis revealed the heterogeneity in MAPK signal activation in CRC organoids, which was associated with diverse responses to EGFR inhibition. Distinct patterns of MAPK pathway activation were observed between CRC organoids sensitive to EGFR-targeted drugs and CRC organoids resistant to EGFR-targeted drugs. This suggests that individual drug efficacy tests for each tumor subpopulation may help improve the prediction of accurate treatment responses in patients.
Based on the clinical response of CRC organoids to chemoradiotherapy and targeted therapy, it is possible for clinicians to identify which patients will benefit from chemoradiotherapy and targeted therapy, so as to customize the treatment plan. Some CRC patients may relapse after receiving chemotherapy. Cho et al 116 established human CRC organoids to explore the related mechanism of 5-fluorouracil resistance, and they found that 5-fluorouracil could activate cancer stem cells in residual lesions through the Wnt/β-catenin pathway. Based on this finding, the combination of Wnt inhibitor and 5-fluorouracil therapy is effective in preventing CRC tumor recurrence. Generally speaking, whole genome sequencing is required before the application of targeted drugs. Only when patients have specific gene mutation targets can they have the indication to use precisely targeted drugs. However, some patients without mutations may also benefit from targeted drugs, so better methods are required to determine the best treatment for patients. Pauli et al 117 used both the CRC organoid model and the xenotransplantation model to test the efficacy of targeted drugs combined with other drugs in the treatment of patients with advanced CRC. The combination of afatinib and vorinostat significantly inhibited the growth of xenograft tumors in mice and reduced the tumor size to only 10% of the size of the tumors in the standard chemotherapy control group. This therapeutic schedule can provide personalized treatment for patients with advanced CRC, bringing new options to patients with advanced CRC. Recently, Narasimhan et al 118 applied organoid technology to guide the clinical development of a precision treatment strategy for CRC patients with peritoneal metastasis and eventually achieved satisfactory outcomes. In summary, these studies show that CRC organoids can accurately predict the therapeutic efficacy of patients and have good predictive value. Personalized treatment according to the predicted results can reduce the incidence of drug resistance, avoid overtreatment, and minimize adverse reactions, which has huge clinical application potential and is expected to be applied in prospective clinical trials.
Challenges and Prospects
With the development of organoid technology, some research teams have developed different organoid culture methods. The use of conditioned medium produced from specific cell lines is in a position to reduce the cost of experiments, which inevitably brings problems such as uncontrolled laboratory variation and reproducibility of results. In addition, most current organoid culture systems rely on Matrigel or other tissue-derived matrices. However, the exact composition of Matrigel or other matrices is unclear and there are differences between batches. Matrigel or other matrices are required to be removed when the researchers perform organoid cryopreservation, DNA and RNA extraction, protein extraction, gene editing, and other operations. The dissolution method of Matrigel also affects the proteomics analysis results of organoids. 119 Studies have shown that there are significant differences in the ECM produced from normal colon tissue and CRC tissue in terms of protein composition, vascular network formation, and tumor proliferation, and tumor ECM has the ability to promote tumor vascularization and vascular metabolic transformation. 120 This means that each type of organoid has the optimal ECM for its culture procedure. Drug screening and efficacy testing of organoids requires standardized establishment of organoids to control genetic and epigenetic trait differences in organoids and to ensure the reliability of experimental results. Most importantly, there are no standardized and uniform guidelines for organoid culture nowadays. Organoid standardization is a pivotal problem that we must spare no effort to deal with.
To address these challenges, both scaffold-based organoid culture technologies and scaffold-free organoid culture systems have been developed, by using engineering techniques to synthesize hydrogels and engineered ECM proteins to replace Matrigel and using engineering techniques such as microfluidic and organ microchips to improve organoid culture technologies. For instance, synthetic ECM can be used to establish human small intestinal organoids, improve the repeatability of experimental results, and clarify the mechanical phenomenon of organoid expansion. 121 Decellularized ECM hydrogel derived from gastrointestinal tissue is a good substitute for Matrigel, and the growth and development of gastrointestinal organoids cultured in this hydrogel are even better than the organoids cultured in Matrigel. 122 This decellularized ECM hydrogel enables to mimic the microenvironment of gastrointestinal tissues and can support long-term organoid culture and transplantation. By using microfluidic chip technology, organoid on-a-chips can control organoids from the level of the microenvironment, establish the crosstalk between tissues and multiple organs, reduce the variability and randomness of organoid development, and then solve the problem of unification and standardization in organoid research. Gjorevski et al 123 used a modular synthetic hydrogel network to identify key ECM parameters that control ISC expansion and organoid formation, creating a fully defined organoid culture system. In this system, the synthetic hydrogels consist of a mixture of type I collagen and Matrigel, which are integrated into a permeable platform to form a hybrid microchip system. The high-hardness matrix enables ISCs to maintain expansion through a YAP-dependent mechanism, while the adhesion of soft matrix and laminin promotes ISC differentiation and organogenesis. This fully defined culture system with a minimal environment can meet the dynamic needs of tissue development, homeostasis, and repair. By using a hybrid matrix composed of a mixture of type-I collagen and Matrigel, Nikolaev et al 124 designed a special scaffold microchip that could be permeable to gases, nutrients, and macromolecules, that would guide ISCs to form tube-shaped epithelial organoids with an accessible lumen, crypt, and villus spatial structure similar to intestine tissue in-vivo. After being connected to an external pumping system, this tubular epithelial organoid can be maintained stably for more than several weeks. It contains rare cell types in common intestinal organoids, such as M cells and intestinal endocrine cells. What's more, this tubular organoid has cell diversity, key physiological characteristics, and strong regenerative ability comparable to the human gut (Figure 4(A)). Embedding ISCs into the softening ECM such as Matrigel would result in uncontrolled budding, a dilemma faced by conventional organoid cultures. Recently, Gjorevski et al 125 developed another standardized approach that enables to specify the organoid geometry to establish intestinal organoids of defined shape, size, and cell distributions, forming structures that are predictable, more similar to normal intestine and reproducible, achieving spatially and temporally control organoid formation. The ISCs were embedded in a synthetic hydrogel containing photosensitive polyethylene glycol. The localized matrix was softened by light treatment at 405 nm wavelength to achieve spatially and temporally specific control of the localized matrix, thus precisely controlling the size and characteristics of intestinal organoids (Figure 4(B)). This study also revealed the key molecular mechanism of crypt-villus axis formation in the organoids, providing profound insights into the control of the size and shape of intestinal organoids, as well as a unified reference for the breaking of intestinal symmetry and the formation of intestinal schemata. In conclusion, tissue engineering can help solve the standardization problems facing current organoid systems. Uniform and controllable culture standards of organoids can further promote the development of organoid research.

The standardization of organoid culture and human intestinal organoid system with human immune cells. (A) Long-term homeostatic culture of tubular mini-guts established by Nikolaev et al 124 via using tissue engineering technology. Brightfield and LGR5–eGFP fluorescence time-course experiments of epithelium formation in tissue-engineered mini-guts compared to traditional organoids formed in Matrigel. From top to bottom, day 0, day 1, day 3, and day 5 of organoid culture, respectively. Scale bars, 50 μm. (B) Organoid formation in spatially and temporally control through photopatterning by Gjorevski et al. 125 Spatially defined crypt formation within photopatterned gels 24 h, 48 h, and 72 h after light-induced softening. Lgr5-eGFP expression and proliferation are localized within the buds, extending into the softened regions. Enterocytes (L-FABP stain), Paneth cells (lysozyme stain), and Enteroendocrine cells (ChrA stain) are found in the organoids. Scale bars, 30 μm. (C) Human intestinal organoid system with human immune cells established by Bouffi et al. 126 Formalin-fixed paraffin-embedded sections of transplanted human intestinal organoid at 12, 16, and 20 weeks stained by IHC with anti-human CD45 antibody. Human fetal intestine at 14.7 and 20.7 post-conceptual weeks (PCW) stained, by immunofluorescence, with anti-human CDH1 (E-cadherin) (blue), anti-human CD45 (green) antibodies and DAPI (white). Scale bar, 100 μm.
Moreover, organoid models cannot fully recapitulate the dynamic and complex microenvironment in the human body, and the lack of the integration of major systems such as the vascular system, nervous system, immune system, and endocrine system is the major shortcoming of intestinal organoid models. Despite the above co-culture methods, the current co-culture system is relatively simple, and differences in the ratio of diverse cell types and composition affect the interpretation of experimental results. Furthermore, organoids cannot fully summarize the overall structure and function of organs, and can only simulate part of the process of diseases. 14 CRC organoids could not fully simulate tumor invasion and metastasis. The metabolic wastes produced within the organoids increase with cell proliferation and cannot be excreted smoothly, affecting the activity and growth of the organoids. In addition, tumor tissues in-vivo have unique vascular networks and there are ischemic necrotic zones in tumors due to the different oxygen utilization abilities of tumor tissues in each region, which is part of the crucial reasons affecting the efficacy of anti-tumor drugs in-vivo. The gas-liquid interface culture method is able to solve the problem of gas exchange to a certain extent, but the current organoid model cannot simulate the distribution of blood vessels in the body and establish normal nutrient metabolism. There is close information transmission and interaction between organs and tissues in the human body. The current organoid culture system lacks interaction information and crosstalk between organs. For instance, when organoids are used for drug testing, information on the toxicity of various drugs to vital organs such as the liver, kidney, and heart is not available.
To this end, establishing complex organoid models that integrate multiple organ systems is a potential and feasible approach to solving the above challenges. Firstly, the vascularization of organoids is a significant challenge in current organoids research. To date, the vascularization of some organoid models relies on the use of vascular endothelial cells, but the complete vascular system cannot be formed just by endothelial cell culture. Holloway et al 127 found endothelial cell population in the early stage of intestinal organoid culture derived from PSCs, while the number of endothelial cells would gradually decrease with the increase of culture time. When cytokines such as EGF, VEGF, bFGF, and BMP4 were added into the medium, endothelial cells could be induced to differentiate into vascularized small intestinal organoids. Recently, several research teams have built a vascularization or vascular perfusion intestinal organoid model with the help of microfluidic chips.128,129 Nashimoto et al 130 integrated cancer cells, umbilical vein endothelial cells, and fibroblasts into microfluidic devices to create vascularized tumor organoids. However, most of the blood vessels only generate around the organoids and do not completely penetrate into the cancer cells to form a network of blood vessels. There are still some limitations in the vascularization of organoids created by microfluidic technology, and the vascularization of organoids needs further study. In 2022, Enrico et al 131 provided a brand new idea for the realization of the organoid model with a complex microvascular system. Femtosecond laser was used to irradiate hydrogel to create stable 3D microchannels with a diameter of 20 to 60 microns, and then artificial microvessels could be formed by injecting 3D microchannels into endothelial cell culture or cell culture medium. More crucially, such 3D microchannels can be created in hydrogels containing organoids and cells without affecting the activity of the extraluminal material.
Immune-epithelial cell crosstalk is vital to the maintenance of intestinal homeostasis, immune tolerance to commensal microorganisms, and defense against gastrointestinal harmful pathogens. Co-culture of patient-derived organoids and immune cells in the same culture dish is the protocol of most studies, but this does not mimic the complex immune response of immune cells in-vivo. Moreover, the number of tumor-associated lymphocytes decreases over time in organoid culture and they cannot be retained for more than 2 months. 85 However, lymphocytes are the target of targeted drugs and immune checkpoint inhibitors used in clinical treatment. Thus, the immune system is a significant integral part of the integration of multisystem organoids for CRC organoids and normal intestinal organoids. Interestingly, Bouffi et al 126 developed the first human intestinal organoids containing immune cells recently. Human intestinal organoids derived from PSCs were transplanted under the kidney capsule of mice with a humanized immune system. After 12, 16, and 20 weeks, the authors found that human CD45 + immune cells migrated to the mucosal layer and formed cellular aggregates that are similar to adult gut lymphoid follicles. When the transplanted human intestinal organoids were exposed to microorganisms, immune cells were activated and then plasma cells were activated to secrete IgA antibodies to neutralize the microorganisms (Figure 4(C)). This human intestinal organoid system with human immune cells provides a framework for future research on intestinal infectious diseases, intestinal allergic, autoimmune, and inflammatory diseases.
In short, organoids that integrate multiple organs are of great importance in drug screening and individualized therapy. Engineering technologies such as organoid-on-a-chips and microfluidic may be effective measures to address these challenges.132,133 Future studies need to more comprehensively simulate the interaction between tumor microenvironment and organs, and further optimize organoid models.
Conclusion
The CRC organoid models have some limitations and there are still some key challenges to overcome in terms of clinical translational applications, but CRC organoid models still have significant advantages, potential, and application value compared with the traditional in-vitro model. With the development of standardized technology such as microfluidic chips, tissue engineering, and air-liquid interactive culture systems, the complexity and accuracy of organoid models will continue to improve. In addition, with the progress of organoid technology, the readings from the organoid model will continue to increase, and the tumor organoid research represented by CRC will also enter a new stage. Accordingly, the application of the CRC organoid model can also promote the optimization of the clinical treatment mode of CRC, which makes CRC really enter the era of individualized treatment.
Footnotes
Acknowledgments
We thank the funders for their support.
Author Contributions
Literature analysis and conceptualization: JW L, JH L, WH S, and H C; original draft preparation and writing: JW L and JH L; artwork: JW L; review and supervision: JH L, WZ X, HW Y, WH S, and H C. All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The National Natural Science Foundation of China (82171698, 82170561, 81741067, 81300279), the Natural Science Foundation for Distinguished Young Scholars of Guangdong Province (2021B1515020003), the Climbing Program of Introduced Talents and High-level Hospital Construction Project of Guangdong Provincial People’s Hospital (DFJH201803, KJ012019099, KJ012021143, KY012021183), the Guangdong Basic and Applied Basic Research Foundation (2022A1515012081).