
Abstract
Objective
Tripartite motif-containing protein 11 (TRIM11), an E3 ubiquitin ligase, possesses a pro-tumor property. Still, the detailed functions of TRIM11 remain not well characterized in prostate cancer.
Methods
PC-3 and DU145 prostate cancer cells were transfected with small interfering RNAs (siRNAs) or lentiviruses for TRIM11 deficiency or overexpression, and microRNA-5193 (miR-5193) mimics were utilized for overexpressing miR-5193. Proliferation, apoptosis, migration, and invasion were examined through CCK-8, colony formation, flow cytometry, wound healing, and transwell assays. MAP kinase-extracellular signal-regulated kinase (MEK1/2) and extracellular signal-regulated kinase (ERK)1/2 activities were detected via immunoblotting. Murine xenograft models were established. Interactions of TRIM11 with miR-5193 were demonstrated via dual luciferase reporter.
Results
TRIM11 deficiency or miR-5193 overexpression exerted antiprostate cancer effects through suppression of proliferation, migration, and invasion as well as enhancement of apoptosis in PC-3 and DU145 cells. The mechanisms by which TRIM11 deficiency or miR-5193 overexpression involved the inactivation of MEK1/2 and ERK1/2. miR-5193 downregulated TRIM11 expressions in prostate cancer cells, and their interactions were confirmed. Further, up-regulated miR-5193 weakened the effects of TRIM11 overexpression on enhancing proliferation, migration, invasion, and activity of MEK1/2 and ERK1/2 as well as alleviating apoptosis of prostate cancer cells. In murine xenograft models, TRIM11 upregulation facilitated tumor growth, which was alleviated by miR-5193 overexpression.
Conclusion
These findings described the oncogenic role of TRIM11 in prostate cancer biology, which was post-transcriptionally modulated by miR-5193.
Introduction
Prostate cancer remains the most frequently diagnosed cancer in males, 1 with an estimated 1,414,259 new cases (7.3%) and 375,304 new deaths (3.8%). 2 Patients with localized prostate cancer with low to moderate risk of relapse typically have favorable outcomes with a 99% 10-year overall survival rate if this disease can be detected and treated early. 3 Curative treatment of localized disease comprises active surveillance, radical prostatectomy as well as ablative radiotherapy. 4 Treatment for recurrent patients following prostatectomy contains salvage radiotherapy and/or androgen deprivation therapy (ADT) against local recurrence, or with ADT plus chemotherapy or new androgen signaling-targeted drugs against systemic recurrence.5,6 Nevertheless, most cases ultimately experience resistance. 7 Hence, it is required for discovering novel treatments that are capable of targeting prostate cancer cells as well as reducing the influence on healthy tissues. In-depth comprehension of additional molecular alterations during prostate cancer progression is essential for the development of novel targets and therapeutics to prolong patients’ survival.
Tripartite motif-containing protein 11 (TRIM11) is an E3 ubiquitin ligase, which belongs to the tripartite motif (TRIM) protein family. 8 The carcinogenic properties of TRIM11 have been characterized in previous studies. For instance, TRIM11 heightens proliferative capacity along with glycolytic process for breast carcinoma cells through modulating AKT/glucose transporter type 1 (GLUT1) signaling 9 or stabilizing estrogen receptor α. 10 Both in gastric cancer 11 and lung cancer, 12 TRIM11 strengthens tumor growth and metastases through activating β-catenin signaling or dual-specificity phosphatase 6 (DUSP6)-mediated extracellular signal-regulated kinase (ERK)1/2 signaling. 13 TRIM11 is capable of promoting proliferation, migration as well as chemoresistance in anaplastic thyroid cancer. 14 TRIM11 inhibition stimulates apoptosis and alleviates cervical cancer cellular progression by phosphoinositide 3-kinase/AKT signaling. 15 Recent research has unveiled that TRIM11 weakens ferritinophagy as well as gemcitabine sensitivity via ubiquitin-conjugating enzyme E2N (UBE2N)/ubiquitin-binding protein Tax1 binding protein1 (TAX1BP1) pathway in pancreatic ductal adenocarcinoma. 16 More importantly, a recent report conducted by Guo et al has indicated that knocking-down TRIM11 can enhance paclitaxel sensitivity to prostate cancer cells. 17 However, how TRIM11 affects the occurrence and development of prostate cancer is still unclear.
Bioinformatics analysis has indicated that TRIM11 could be post-transcriptionally modulated by microRNAs to modulate the progression of numerous cancers. For example, TRIM11 serves as a target of miR-24-3p to induce the proliferative potentials of colon cancer, breast cancer, and mantle cell lymphoma, respectively.18‐20 Our previous study has indicated that TRIM11 is overexpressed in prostate cancer and its expression can be downregulated by microRNA-5193 (miR-5193). 21 Interestingly, another study conducted by Song et al 22 also demonstrated that miR-5193 is an important regulator of TRIM11 to function as a tumor suppressor in the development of ovarian cancer. It was capable of weakening proliferative and migratory capacities of ovarian cancer cells through post-transcriptionally modulating TRIM11. 22 Still, the mechanisms of TRIM11 and miR-5193 in prostate cancer remain not well characterized. In our work, we hypothesize that TRIM11 may be also post-transcriptionally modulated by miR-5193, and therefore affects the progression of prostate cancer.
Materials and Methods
The reporting of this study conforms to ARRIVE 2.0 guidelines. 23
Cell Culture
Human prostate cancer cell lines PC3 and DU145 (ATCC, USA) were maintained in Dulbecco's modified Eagle medium (DMEM) (Gibco, USA) plus 10% fetal bovine serum along with 1% penicillin–streptomycin in a humidified environment of 5% CO2/95% air at 37 °C.
RNA Interference
PC3 or DU145 cell line was seeded into six-well plates (1 × 105 cells per well). When the confluence reached 75%, the cells were transfected with negative control of small interfering RNA (siRNA) (si-NC), si-TRIM11-395, si-TRIM11-725, si-TRIM11-1214, miR-5193 mimics or NC mimics utilizing Lipofectamine 2000 (Invitrogen). The sequences included: si-NC: 5′-UUCUCCGAACGUGUCACGUTT-3′, 5′-ACGUGACACGUUCGGAGAATT-3′; si-TRIM11-395: 5′-CCGAAGACCUCAAGGCGAATT-3′, 5′-UUCGCCUUGAGGUCUUCGGTT-3′; si-TRIM11-725, 5′-GGCUGCUGCAGGACAUCAATT-3′, 5′-UUGAUGUCCUGCAGCAGCCTT-3′; si-TRIM11-1214, 5′-GGAUCUUUCUGGACUACGATT-3′, 5′-UCGUAGUCCAGAAAGAUCCTT-3′.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA extraction was implemented with Trizol (10606ES60; Yeasen), followed by synthesis of cDNA with cDNA Synthesis SuperMix (11123ES60; Yeasen). Quantification of the mRNA expressions was conducted with 12K quantitative real-time polymerase chain reaction (RT-qPCR) instrument (ABI) and qPCR SYBR Green Master Mix (11202ES08; Yeasen). Primer sequences included: TRIM11, 5′-GGACGCAATGCTGTTCCAAG-3′, CAAACTCGCCCAGCACATTC-3′; GAPDH, 5′-TCAAGAAGGTGGTGAAGCAGG-3′, 5′-TCAAAGGTGGAGGAGTGGGT-3′; miR-5193, 5′-AGTGCAGGGTCCGAGGTATT-3′, 5′-CGCGTCCTCCTCTACCTCAT-3′; U6, 5′-GCTTCGGCAGCACATATACTAAAAT-3′, 5′-CGCTTCACGAATTTGCGTGTCAT-3′.
Cell Viability Assay
PC3 or DU145 cell line (5000/100 μl) was planted onto 96-well plates. At indicated times, 10 μl cell counting kit-8 (CCK8; CK04; Dojindo) was mixed with the medium. Following 1-h incubation, PC3 or DU145 cells were exposed to 10 μl stop buffer. Cell viability was evaluated via examining the absorbance at 450 nm.
Flow Cytometry
Flow cytometry was implemented via apoptosis Detection Kit (AD10; Dojindo). PC3 or DU145 cell line was washed with PBS, along with 0.25% trypsin digestion for 3 min. Afterwards, they were centrifuged at 1000 rpm for 5 min, and the supernatant was discarded. 1 × Annexin V binding reagent was utilized for preparing cellular suspension with 1 × 106 cells/ml. Incubation of 5 μl Annexin V-FITC conjugate with 100 μl cellular suspension was implemented for 10 min away from the light. Thereafter, incubation with 5 μl of PI was conducted for 10 min away from the light, followed by 400 μl of 1 × Annexin V binding reagent. Within 1 h, apoptosis was examined through flow cytometer.
Colony Formation Assay
PC3 or DU145 cell line was seeded into six-well plates at 2000 cells per well. DMEM was exchanged other day. Colonies were counted following 2 weeks. After discarding the supernatant and washing, fixation was conducted with 4% paraformaldehyde (P0099; Beyotime) for 15 min. The fixative was discarded as well as 0.1% crystal violet (C0121; Beyotime) staining was achieved for 15 min. After washing with PBS, number of colonies was counted and photographed utilizing an IX71 inverted phase contrast microscope (Olympus).
Wound Healing Assay
With a marker, horizontal lines were drawn on the back of the 6-well plate evenly, about every 0.5–1 cm, across the holes. PC3 or DU145 cells were plated in six-well plates, adhered overnight, and the cell density was controlled to reach 100% on the second day. The next day, scratches were made on the back utilizing 200 μl pipette tips. Following the removal of the streaked cells, serum-free DMEM was added. At 0 h along with 24 h, photographs were taken with an IX71 inverted phase contrast microscope (Olympus).
Transwell Assay
Matrigel (354234; BD) was diluted 40-fold with pre-chilled medium. In total, 100 μL of Matrigel was added to the invasion chamber that was placed in a 24-well plate for 30 min. PC3 or DU145 cells were adjusted to 1 × 106 cells/ml. In total, 100 μL cellular suspensions were added onto the upper chamber as well as 600 μL serum-containing DMEM was added onto the lower chamber. The chamber was taken out after 24 h. The cells were fixed with 4% paraformaldehyde (P0099; Beyotime) for 15 min, along with 0.1% crystal violet (C0121; Beyotime) for 15 min. With cotton swabs, removal of unpenetrated cells within the filter surface was conducted. Photographs were acquired with an IX71 inverted phase contrast microscope (Olympus).
Immunoblotting
PC3 or DU145 cells were lysed by RIPA lysis buffer (BL504A; Biosharp) on ice for 15 min. Cell lysates were harvested and centrifuged at 12,000 rpm at 4 °C for 5 min. The supernatant was transferred to a new 1.5 ml EP tube. In total, 3 µl of BCA kit (BL521A; Biosharp) was applied for examining total protein content. 5 × Loading Buffer was added, followed by heat at 100 °C for 10 min. 12% SDS polyacrylamide separating gel was prepared. The protein was transferred onto PVDF membrane, which was sealed with 5% milk/TBST for 1 h. Primary antibody including TRIM11 (cat.no. ab111694; Abcam; 1:1000), ERK1/2 (cat.no. 16443-1-AP; Cell signaling; 1:1000), p-ERK1/2 (cat.no. 28733-1-AP; Proteintech; 1:2000), MAP kinase-extracellular signal-regulated kinase (MEK)1/2 (cat.no. 8727T; Cell signaling; 1:1000), p-MEK1/2 (cat.no. 9154T; Cell signaling; 1:1000), GAPDH (cat.no. 60004-1-Ig; Proteintech; 1:10000) was diluted with 1% BSA/PBST, and the membrane was sealed by hybridization bag, and cooled at 4 °C overnight. The membrane was placed in horseradish peroxidase-labeled goat anti-rabbit or anti-mouse secondary antibody (cat.no. A21020 or A21010; Abbkine; 1:10000) diluted with 5% milk/PBST, along with incubation for 1 h. The protein bands were developed with ECL luminescent fluid and analyzed with fully automatic chemiluminescence imaging system (5200Multi; Tanon).
Lentivirus Construction and Transfection
Virus coating was performed using psPAX2 and pMD2.G plasmids, and pMD2.G, psPAX2 and pCDH-TRIM11 plasmids were mixed in a mass ratio of 1:3:4. 293T cells were cultured in 10 cm dishes, and transfection was carried out when the cells reached 70% confluence. In total, 20 μg of plasmids were used per dish, and 20 μl Lipofectamine 2000 (Invitrogen) was used for transfection. Serum-free DMEM with no double antibody was used for culture. After 6 h, it was replaced with DMEM without double antibody and serum to continue the culture. The cell culture medium was collected at 24 and 48 h after transfection, filtered with a 0.2 μm filter, and directly used to culture PC3 cells. PC3 cells were cultured in 6-well plates, 2 mL of virus-containing cell culture medium was added, along with 10 ug/mL polybrene. After 24 h, the cells were replaced with fresh DMEM without double antibody and serum to continue the culture. In total, 48 h after infection, 2 μg/mL puromycin was added for screening. After 2 weeks of culture, transfection effects were evaluated with RT-qPCR.
Dual Luciferase Reporter
The 3′UTR wild-type (TRIM11-wt) along with mutant (TRIM11-mut) sequences of TRIM11 gene were synthesized and incorporated into fluorescent reporter plasmids, respectively. Transfection of luciferase reporter plasmid along with miR-5193 mimics or NC mimics into 293 T cells was implemented via Lipofectamine 2000 reagent (Invitrogen). The fluorescence activity was detected 48 h later utilizing Dual-Luciferase Reporter Assay System (Promega).
Xenograft Tumor Model
Fifteen male nude mice weighting 20 to 24 g (4 weeks; Charles River Laboratories) were randomized to control group, TRIM11-overexpressed (OE-TRIM11) + NC mimics group as well as OE-TRIM11 + miR-5193 mimics group (n = 5). All mice were raised in a specific pathogen-free environment for one week. A total of 0.2 mL cell suspensions (5 × 106) of normal PC3 cells or PC3 cells stably transfected with OE-TRIM11 were planted subcutaneously on the side of the back of nude mice. 24 When the tumors grew to the size of rice grains after 2 weeks, miR-5193 mimics or PBS (K813; Shanghai Gao Chuang) was injected with equal volumes, once every 3 days, for a total of 8 injections. The tumor volumes (v) were detected as well as recorded every 3 days in accordance with the formula: v = π/6 × short diameter2 × long diameter. The sodium pentobarbital was mixed with sterile saline to make a 3% solution and injected intraperitoneally at a dose of 50 mg/kg (1-1.25 mg per mouse). 25 The mice were sacrificed by cervical dislocation and mortality was confirmed by cessation of heartbeat. The excised tumors were weighed at 4 weeks. The dissected tumors were stored in liquid nitrogen. We followed the Guide for the Care and Use of Laboratory Animals, eighth Edition. 26 During this experiment, we make efforts to minimize the number of animals utilized and to decrease their suffering. Animal experiments were approved by the animal ethics committee of our hospital (approval number: WYYY-AEC-2020-063; Signature date: 7 December 2020).
Statistical Analysis
In vitro experiments were performed in triplicate, and each experiment was repeated three times. In vivo experiments were performed using six mice per group. Statistical analysis was implemented with GraphPad Prism 8.0.1 (LaJolla, CA, USA). Measurement data are displayed as mean ± standard deviation. Student's t-tests or one-way analyses of variance were applied. Statistical significance was set at P < .05.
Results
TRIM11 Deficiency Induces Apoptosis as Well as Impairs Proliferative Capacity in Prostate Cancer Cells
The oncogenic role of TRIM11 in prostate carcinoma biology was probed in two prostate cancer cell lines, PC-3 along with DU145. TRIM11 knockout cell lines were produced through siRNA transfections. In Figure 1A, si-NC did not affect TRIM11 mRNA expressions than blank cells. Three TRIM11 siRNAs (si-TRIM11-395, si-TRIM11-725, or si-TRIM11-1214) significantly lowered TRIM11 mRNA expressions than si-NC. Si-TRIM11-395 presented the optimal knockout effect on TRIM11 mRNA expressions, which was applied for subsequent assays. CCK-8 was carried out for determining whether TRIM11 was required for prostate cancer progression. Cell viability of PC-3 and DU145 was significantly restrained at 24, 36, 48, and 72 h following TRIM11 expressions were knocked out (Figure 1B and C). Higher apoptosis rate was noted in TRIM11-depleted prostate cells (Figure 1D to F). Additionally, TRIM11 depletion impaired clone formation ability of PC-3 or DU145 cell line (Figure 1G to I).

TRIM11 deficiency induces apoptosis and impairs proliferative capacity in prostate cancer cells. (A) Transfection effect of si-TRIM11-395, si-TRIM11-725 or si-TRIM11-1214 on TRIM11 mRNA expressions in PC-3 cells via RT-qPCR. (B, C) Cell viability of PC-3 or DU145 cell line with si-NC or si-TRIM11 transfections. (D-F) Apoptosis of PC-3 or DU145 cell line with si-NC or si-TRIM11 transfections. (G-I) Number of colonies of PC-3 or DU145 cell line with si-NC or si-TRIM11 transfections. *P < .05; **P < .01; ***P < .001; ****P < .0001. Abbreviations: TRIM11, tripartite motif-containing protein 11; NC, negative control; RT-qPCR, quantitative real-time polymerase chain reaction.
TRIM11 Loss Restrains the Motility and Invasiveness of Prostate Carcinoma
The migratory along with invasive capacities of tumors allow them to spread with the tissues and metastasize to distant organs. 27 For investigating the role of TRIM11 in prostate cancer metastases, prostate cancer cellular migration and invasion were estimated via wound healing or Transwell experiment. In Figure 2A to C, migratory capacities of PC-3 or DU145 cell line were significantly impaired via TRIM11 loss. Additionally, depleted TRIM11 in prostate cells displayed dramatic reduction in invasive capacities (Figure 2D to F).
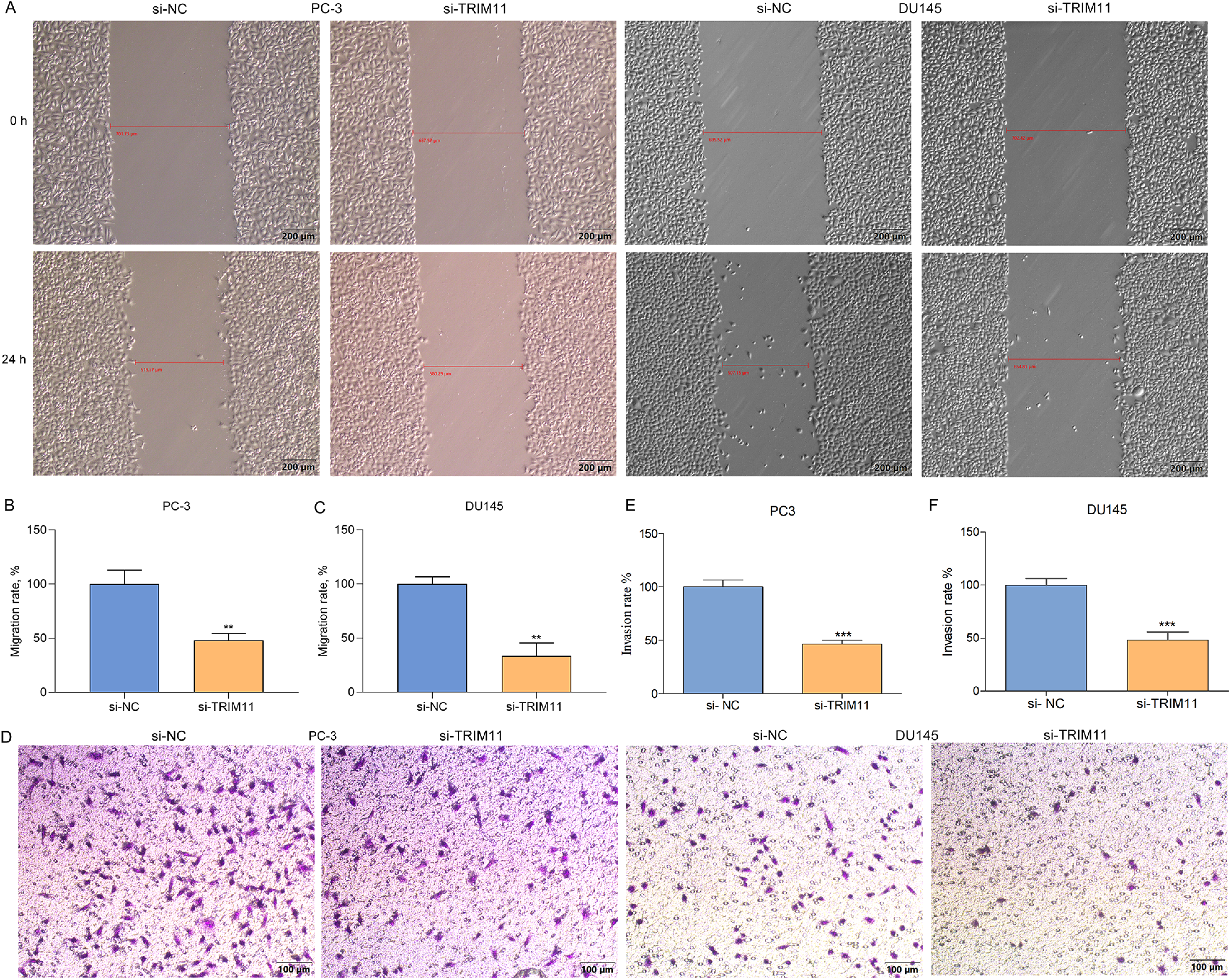
TRIM11 loss restrains the motility and invasiveness of prostate cancer cells. (A-C) Photographs and quantification for wound healing experiment of TRIM11-depleted PC-3 or DU145 cell line at 0 and 24 h. Scale bar: 200 μm. (D-F) Photographs and quantification for transwell invasion experiment of TRIM11-depleted PC-3 or DU145 cell line. Scale bar: 100 μm. **P < .01; ***P < .001.
TRIM11 Mediates MEK/ERK Pathway in Prostate Cancer
Targeting MEK/ERK pathway is a viable treatment strategy against prostate cancer. 13 Limited evidence indicates that TRIM11 mediates MEK/ERK pathway in diabetic nephropathy 28 and lung cancer. 13 Immunostaining results showed that TRIM11 depletion did not affect ERK1/2 or MEK1/2, but remarkably decreased phosphorylated ERK1/2 and MEK1/2 both in PC-3 along with DU145 cell lines (Figure 3A to F). This indicated that TRIM11 mediated MEK / ERK signaling activation in prostate cancer.
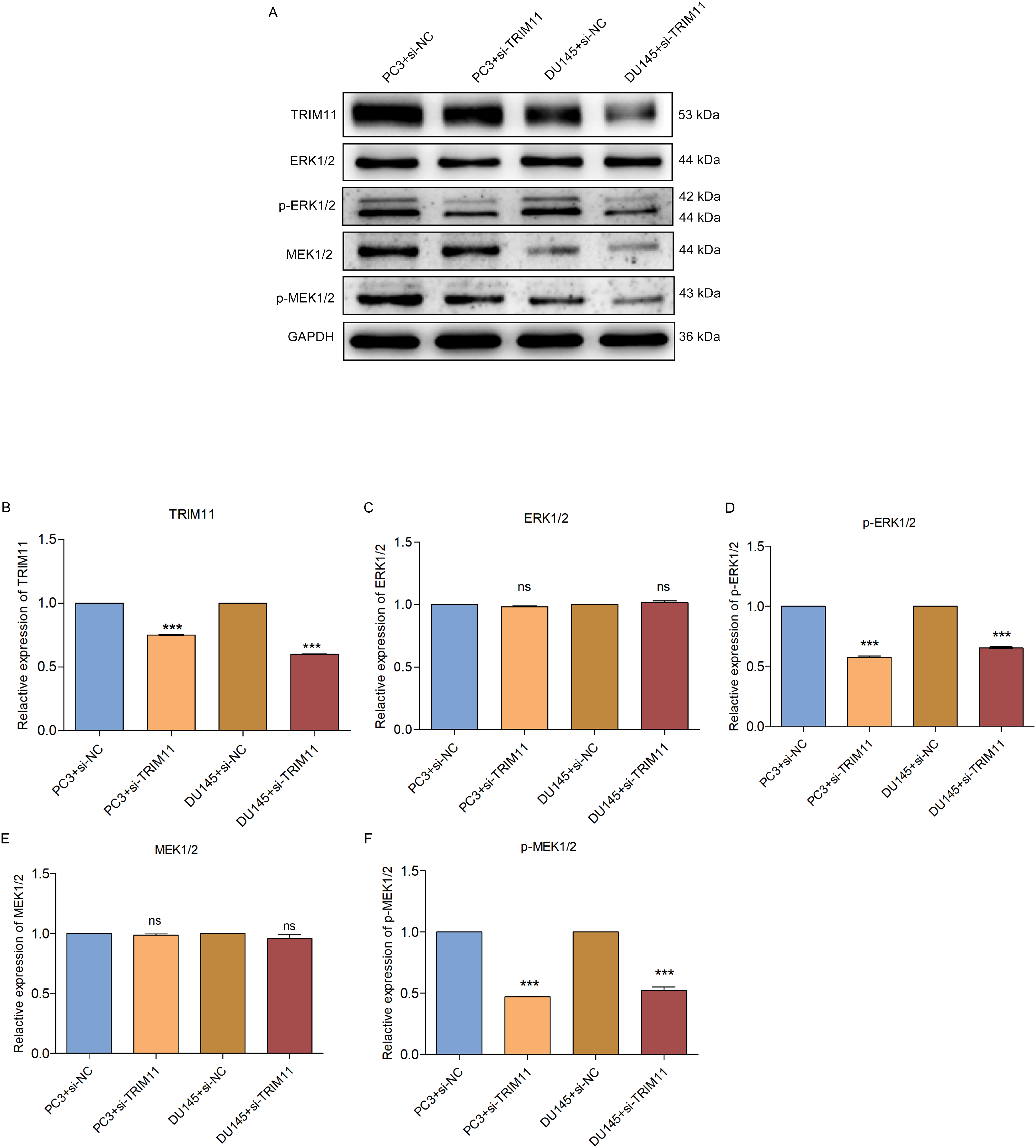
TRIM11 mediates MEK/ERK pathway in prostate cancer. (A-F) Immunoblotting of TRIM11, ERK1/2, p-ERK1/2, MEK1/2, and p-MEK1/2 in TRIM11-depleted PC-3 or DU145 cell line. Ns: no significance; ***P < .001. Abbreviations: ERK, extracellular signal-regulated kinase; MEK, MAP kinase-extracellular signal-regulated kinase; OE-TRIM11, tripartite motif-containing protein 11-overexpressed.
MiR-5193 Overexpression Represses Cell Growth, Migration and Invasion of Prostate Cancer Cells
Limited evidence indicates that TRIM11 is post-transcriptionally modulated by miR-5193. 22 Herein, we further probed the function of miR-5193 in prostate cancer biology. MiR-5193 expressions were dramatically increased in PC-3 or DU145 cell line with miR-5193 mimics than NC-mimics (Figure 4A and B). In Figure 4C and D, miR-5193-overexpressed PC-3 or DU145 cell line presented significantly lower viability. Moreover, apoptosis of PC-3 or DU145 cell line was significantly heightened through miR-5193 mimics (Figure 4E to G). As shown in Figure 4H to J, clone formation abilities of two prostate cancer cell lines were impaired by miR-5193 mimics. Additionally, miR-5193-overexpressed PC-3 or DU145 cell line presented attenuated migratory (Figure 4K to M) as well as invasive capacities (Figure 4N to P).

MiR-5193 overexpression represses cell growth, migration and invasion of prostate cancer cells. (A, B) MiR-5193 expressions in PC-3 or DU145 cell line with miR-5193 mimics. (C, D) Cell viability of PC-3 or DU145 cell line with miR-5193 mimics. (E-G) Apoptosis rate of PC-3 or DU145 cell line with miR-5193 mimics. (H-J) Number of colonies of PC-3 or DU145 cell line with miR-5193 mimics. (K-M) Wound healing for the migratory capacity of PC-3 or DU145 cell line with miR-5193 mimics. Scale bar: 200 μm. (N-P) Transwell invasion experiment for the invasive capacity of PC-3 or DU145 cell line with miR-5193 mimics. Scale bar: 100 μm. *P < .05; **P < .01; ***P < .001; ****p < .0001. Abbreviations: miR-5193, microRNA-5193; OE-TRIM11, tripartite motif-containing protein 11-overexpressed.
MiR-5193 post-Transcriptionally Modulates TRIM11 and Attenuates MEK/ERK Signaling Activation in Prostate Cancer Cells
Further analysis showed that TRIM11 expressions were significantly lower in miR-5193 mimics-transfected PC-3 or DU145 cell line (Figure 5A to D). Additionally, miR-5193 mimics did not alter ERK1/2 or MEK1/2 but decreased phosphorylated ERK1/2 and MEK1/2 in PC-3 or DU145 cell line (Figure 5E to H), demonstrating that miR-5193 attenuated MEK/ERK signaling activation. Dual luciferase reporter showed that fluorescence activity was dramatically decreased under transfection with TRIM11-wt and miR-5193 mimics (Figure 5I). Nevertheless, no significant difference in fluorescence activity was investigated when co-transfection with TRIM11-mut and miR-5193 mimics. Thus, TRIM11 can be post-transcriptionally modulated by miR-5193.
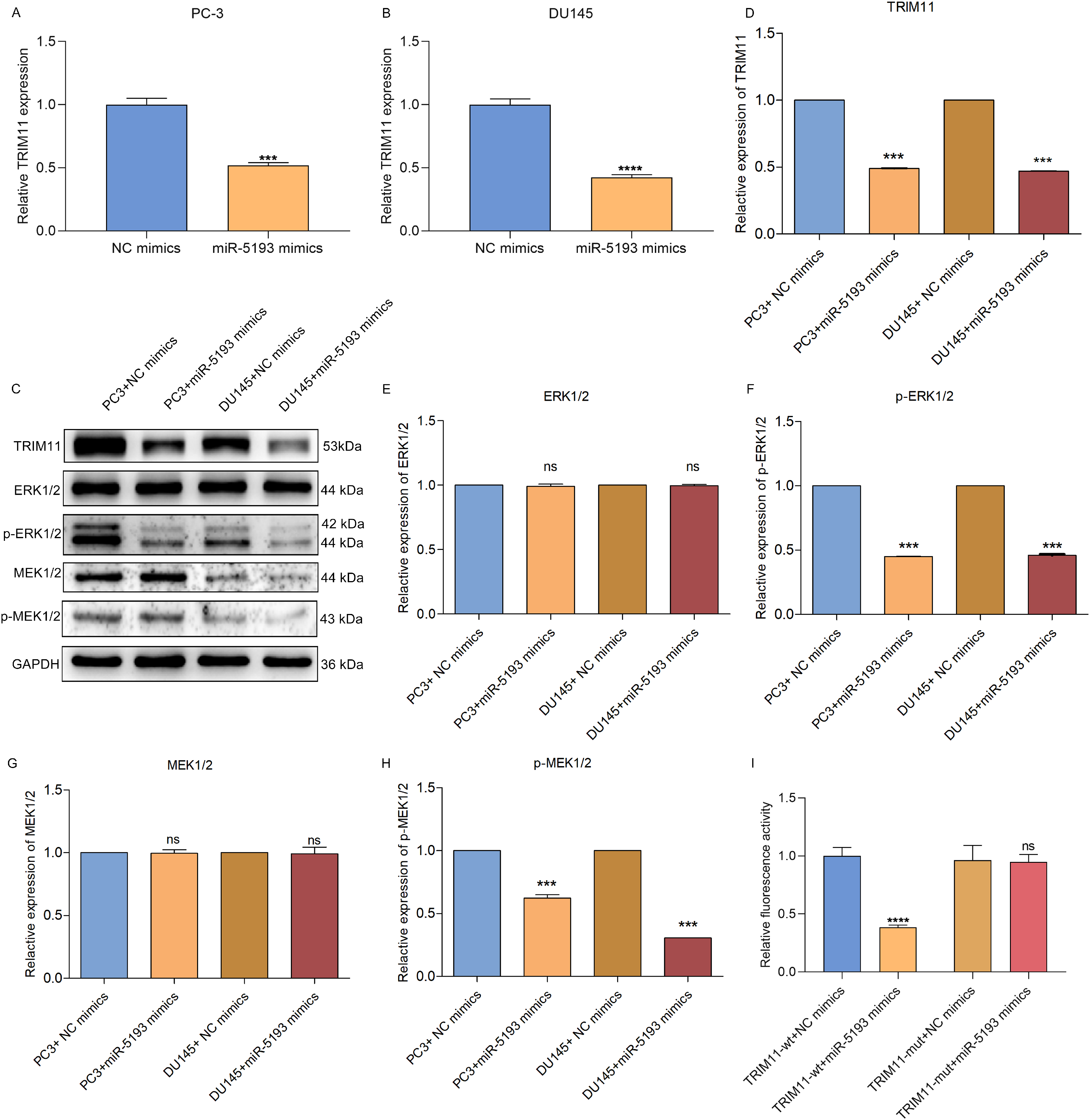
MiR-5193 post-transcriptionally modulates TRIM11 and attenuates MEK/ERK signaling activation in prostate cancer cells. (A, B) TRIM11 mRNA expressions of PC-3 or DU145 cells with miR-5193 mimics. (C-H) Immunoblotting of TRIM11, ERK1/2, p-ERK1/2, MEK1/2, and p-MEK1/2 in PC-3 or DU145 cell line with miR-5193 mimics. (I) Dual luciferase reporter between TRIM11-wt or TRIM11-mut and miR-5193. Ns: no significance; ***p < .001; ****p < .0001. Abbreviations: ERK, extracellular signal-regulated kinase; MEK: MAP kinase-ERK kinase; miR-5193, microRNA-5193; OE-TRIM11, tripartite motif-containing protein 11-overexpressed; wt, wild-type; mut, mutant.
Overexpression of miR-5193 Prohibits Prostate Cancer Cell Growth, Migration and Invasion Through TRIM11
We further probed the underlying mechanisms by which TRIM11 and miR-5193 participated in prostate cancer progression and metastases. We firstly overexpressed TRIM11 in PC3 cells (Figure 6A). Overexpressed TRIM11 facilitated viability of PC3 cells, weakened via miR-5193 mimics (Figure 6B). In Figure 6C and D, PC3 cellular apoptosis was alleviated via TRIM11, which was reversed by miR-5193 mimics. In agreement, overexpressed TRIM11 heightened colony formation capacity of PC3 cells, weakened through miR-5193 mimics (Figure 6E and F). Moreover, PC3 cellular migration and invasion were heightened via overexpressed TRIM11, which were weakened by miR-5193 mimics (Figure 6G to J). Overall, miR-5193 facilitated prostate cancer cell growth and metastases through TRIM11.
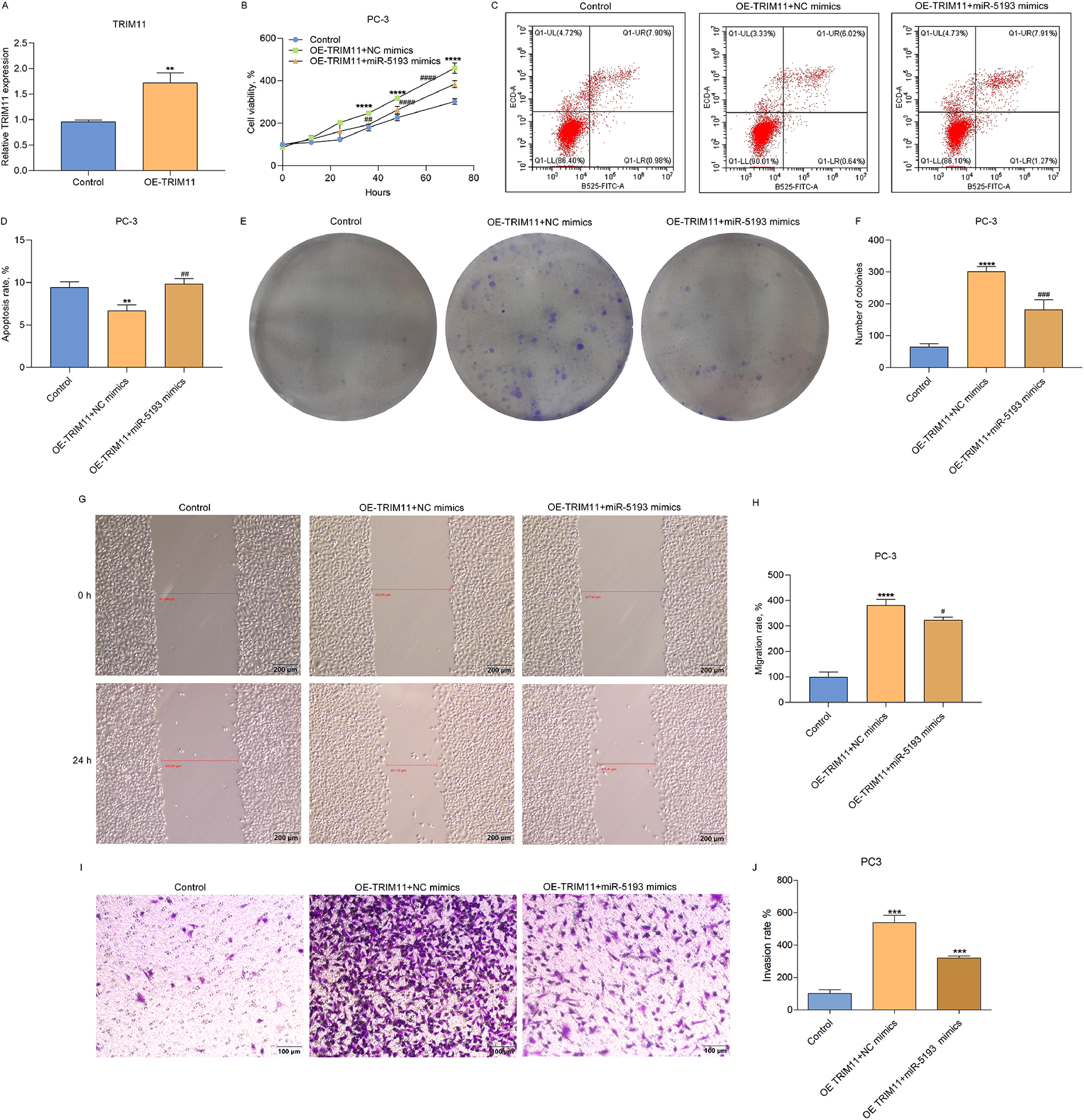
Overexpression of miR-5193 prohibits prostate cancer cell growth, migration and invasion through TRIM11. (A) TRIM11 mRNA expressions in PC-3 cells with TRIM11-overexpressed (OE-TRIM11). (B) Cellular viability, (C, D) apoptotic level, (E, F) number of colonies, (G, H) migratory capacity, (I, J) invasive capacity of control-, TRIM11-overexpressed (OE-TRIM11) + NC mimics or OE-TRIM11 + miR-5193 mimics-PC3 cells. Scale bar: 200 μm (Wound healing assay); Scale bar: 100 μm (Transwell assay). Compared with control, **P < .01; ****P < .0001. Compared with OE-TRIM11 + NC mimics, #P < .05; ##P < .01; ###P < .001; ####P < .0001. Abbreviations: miR-5193, microRNA-5193; OE-TRIM11, tripartite motif-containing protein 11-overexpressed; NC, negative control.
MiR-5193 Attenuates MEK/ERK Signaling Activation Through TRIM11
In TRIM11-overexpressed PC-3 cells, miR-5193 mimics decreased TRIM11 expressions (Figure 7A and B). Overexpressed TRIM11 did not alter ERK1/2 or MEK1/2 but increased phosphorylated ERK1/2 and MEK1/2 in PC-3 cells (Figure 7C to F). Oppositely, miR-5193 mimics alleviated the increase in phosphorylated ERK1/2 and MEK1/2 in TRIM11-overexpressed PC-3 cells. MiR-5193 attenuated MEK/ERK signaling activation through TRIM11 in prostate carcinoma cells.

miR-5193 attenuates MEK/ERK signaling activation through TRIM11. (A-F) Immunoblotting of TRIM11, ERK1/2, p-ERK1/2, MEK1/2, and p-MEK1/2 in control-, TRIM11-overexpressed (OE-TRIM11) + NC mimics or OE-TRIM11 + miR-5193 mimics-PC3 cells. Compared with control, ***P < .001; compared with OE-TRIM11 + NC mimics, ####P < .0001; ns: no significance. Abbreviations: ERK, extracellular signal-regulated kinase; MEK, MAP kinase-ERK kinase; miR-5193, microRNA-5193; OE-TRIM11, tripartite motif-containing protein 11-overexpressed; NC, negative control.
MiR-5193 Alleviates Tumor Growth in Vivo Through TRIM11
Murine xenograft models were set up utilizing control-, TRIM11-overexpressed (OE-TRIM11) + NC mimics- or OE-TRIM11 + miR-5193 mimics-PC3 cells. After 21 days, the xenografts were collected. Xenografts formed by OE-TRIM11 + NC mimics-PC3 cells were bigger than control cells (Figure 8A and B). Oppositely, OE-TRIM11 + miR-5193 mimics group had a smaller tumor volume than OE-TRIM11 + NC mimics group. The tumor growth curve indicated that tumors in OE-TRIM11 + NC mimics group grew faster than the control group (Figure 8C). In comparison to OE-TRIM11 + NC mimics group, tumor growth was slower in OE-TRIM11 + miR-5193 mimics group. The xenografts were also weighed. In Figure 8D, xenografts formed by OE-TRIM11 + NC mimics-PC3 cells were heavier in comparison to control cells. Compared with OE-TRIM11 + NC mimics group, smaller tumor weigh was found in OE-TRIM11 + miR-5193 mimics group. Hence, miR-5193 enabled to alleviate tumor growth in vivo through TRIM11. Thereafter, we examined miR-5193 and TRIM11 expressions in xenografts. Lower miR-5193 and higher TRIM11 expressions were found in OE-TRIM11 + NC mimics group than the control group (Figure 8E and F). Oppositely, compared with OE-TRIM11 + NC mimics group, miR-5193 expressions were up-regulated and TRIM11 expressions were down-regulated in OE-TRIM11 + miR-5193 mimics group.

MiR-5193 alleviates tumor growth in vivo through TRIM11. (A, B) Photographs of nude mice and excised subcutaneous tumors through implanting control-, TRIM11-overexpressed (OE-TRIM11) + NC mimics- or OE-TRIM11 + miR-5193 mimics-PC3 cells. (C, D) Quantification of tumor volume and weight. (E, F) miR-5193 and TRIM11 expressions in excised subcutaneous tumors via RT-qPCR. Compared with control, **P < .01; ***P < .001; ****P < .0001. Compared with OE-TRIM11 + NC mimics, ##P < .01; ####P < .0001. Abbreviations: OE-TRIM11, tripartite motif-containing protein 11-overexpressed; miR-5193, microRNA-5193; NC, negative control; RT-qPCR: quantitative real-time polymerase chain reaction.
Discussion
Prostate cancer ranks as the fifth leading cause of cancer-related death among males worldwide. 29 Recently, a significantly increasing incidence and mortality trend was observed in prostate cancer patients in China. 30 The recurrence rate of prostate cancer patients is still high (approximately 40%) although various therapeutic options can successfully cure the majority of patients.31,32 Therefore, exploring the potential molecular targets for prostate cancer treatment is an urgent problem to be solved. This study mainly discussed the underlying functions of TRIM11 in the pathogenesis of prostate cancer, indicating that TRIM11 contributes to the malignancy of prostate cancer via post-transcriptionally modulated by miR-5193.
TRIM11 up-regulations have been reported in prostate cancer more than normal tissues, and correlate to poor outcomes, 21 but its specific function in tumorigenesis remains indistinct. SiRNA is a powerful gene silencing tool that enables selectively suppress specific genes, displaying great potential in gene therapy.33,34 Public data demonstrated that inhibition of TRIM11 dramatically prohibited the proliferative capacities of cells in breast, lung, and ovarian cancers.9,12,35 Consistently, our findings also demonstrated that knockout TRIM11 enabled to alleviate PC3 and DU145 cell proliferation as well as heighten apoptosis. 21 These results implied that knocking-down TRIM11 was helpful for the suppressive effect on cell growth in prostate cancer. Standard therapy of nonmetastatic prostate cancer, to prevent metastasis, remains ADT. 36 Nevertheless, most patients will eventually develop castration-resistant prostate cancer, and thus proves challenging to treat, with a five-year survival of 30%. 37 Impeding prostate cancer metastases enables to ameliorate patients’ prognosis.38,39 Due to cancer metastases as an extremely complex process triggered by diverse crucial events, comprehending the molecular mechanisms underlying these events is beneficial for clinical management.40‐42 Additionally, determining and targeting vital genes involved in prostate cancer metastases is essential to develop future therapeutic regimens to control metastatic spread.3,43,44 Migration and invasion of tumor cells represent two crucial biological phenomena during metastases.45,46 Herein, loss of TRIM11 decreased the migratory and invasive capacities of prostate cancer cells. In line with our experimental data, several researchers have determined that TRIM11 is correlated to migration and invasion in several human cancers. For instances, Lan et al found that TRIM11 triggered migration, invasion as well as EMT of gastric cancer via heightening β-catenin activity. 11 Tang et al 14 detected the pro-migration effect of TRIM11 in anaplastic thyroid cancer. Wang et al 12 also reported that TRIM11 enabled to heighten migration and invasion of lung cancer. Liu et al 15 demonstrated the inhibition effect of TRIM11 deficiency on migration and invasion of cervical cancer. On the basis of these findings, TRIM11 enabled to facilitate the migratory and invasive potentials of prostate cancer cells.
MEK/ERK signaling is a classical signaling pathway for cancer therapy. The cytoplasmic Ser/Thr kinases ERK1 and ERK2 were found to promote cell cycling 47 ; and ERK1/2 activity was shown to be enhanced by yet other cytosolic kinases, MEK1/2, that phosphorylate the conserved Thr/Tyr in the activation loop of ERK1/2. 48 Further investigation of the kinase cascade revealed that cellular RAF gene (CRAF) is the upstream kinase that phosphorylates MEK1 at Ser222 and MEK2 at Ser218 that regulates the activity of MEK, and through which ERK, 49 thus rank-ordering the MAPK signaling from RAS, RAF, MEK, and finally to ERK. 50 In clinical practice, MEK inhibitors including trametinib and cobimetinib have been applied to the treatment of melanoma and non-small-cell lung cancer to limit the metastatic capacities.51,52 In the progression of prostate cancer, phosphorylated ERK1/2 is higher in castration-resistant prostate cancer in comparison to primary patients. 53 Additionally, ERK1/2 phosphorylation is linked to biochemical failure following radical prostatectomy, which is independent of clinicopathologic factors. MEK/ERK pathway heightens androgen receptor element-induced gene transcriptions as well as DNA synthesis of prostate carcinoma. 54 MEK/ERK phosphorylation facilitates prostate tumorigenesis. 55 Hence, we speculated molecules targeting MEK / ERK pathway to limit its activity might be a viable therapeutic strategy against refractory metastatic prostate cancer. As expected, we found that TRIM11 knockdown reduced MEK / ERK signaling activity in prostate cancer. We believed that downregulation of TRIM11 could inactivate MEK / ERK signaling to attenuate prostate cancer progression.
The post-transcriptional regulation of TRIM11 by miRNAs has been uncovered in numerous human cancers such as colon cancer, breast cancer and mantle cell lymphoma.18‐20 Additionally, our previous study in 2019 has confirmed that TRIM11 was a direct target of miR-5193. 21 Meanwhile, Song et al 22 demonstrated that miR-5193 alleviated proliferative and migratory abilities of ovarian cancer cells through targeting TRIM11. Therefore, in the current study, we speculated TRIM11 may be post-transcriptionally regulated by miR-5193 to exert oncogene role in prostate cancer progression. Interestingly, we demonstrated that overexpression of miR-5193 reduced the levels of TRIM11, suggesting that TRIM11 was negatively modulated by miR-5193. These results further implied that miR-5193 could attenuate the influences of TRIM11 overexpression on prostate cancer progression, which was confirmed in our researches, namely upregulation of miR-5193 ameliorating the promoting effects of TRIM11 overexpression on cell growth, migration, and invasion in vitro and tumor growth in vivo. At the same time, miR-5193 upregulation also reversed the activation effects of TRIM11 overexpression on MEK/ERK signaling pathway. We, therefore, concluded that TRIM11 could be post-transcriptionally modulated by miR-5193 and then activate MEK/ERK signaling pathway to facilitate tumor growth and metastasis of prostate cancer.
Some limitations exist in the current study. First, only miR-5193 mimics were used in cell culture, and miR-5193 inhibitor should be added in future studies to make our results more rigorous. Second, in vivo experiments exploring the interactions among TRIM11, miR-5193 and MEK/ERK signaling may be needed. Third, there may be some homology differences between mouse and human. Further clinical experiments are indispensable to validate these findings. Fourth, the levels of up-stream regulatory factors of MEK/ERK signaling such as CRAF, RAS, and RAF should be also tested.
Conclusion
Collectively, TRIM11 was determined as a driver and therapeutic target of prostate cancer, post-transcriptionally modulated by miR-5193. Thus, our study provided preclinical evidence for TRIM11 as a promising therapeutic target for clinical application.
Footnotes
Abbreviations
Acknowledgements
The authors acknowledge the strong support of The First Affiliated Hospital of Wenzhou Medical University.
Ethics Statement
According to the principles of ARRIVE guidelines, Animal experiments were approved by the animal ethics committee of The First Affiliated Hospital of Wenzhou Medical University (Approval No. WYYY-AEC-2020-063; Signature date: 7 December 2020).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.