
Abstract
Introduction
The lethality rate of human lung cancer has ranked near the top in recent years, and lung cancer is regarded as a highly deadly and aggressive tumor, especially nonsmall cell lung cancer (NSCLC).1,2 It has been shown that more than 90% of cancer-related deaths in NSCLC are promoted by tumor progression, especially metastasis. Human lung adenocarcinoma, a major subtype of NSCLC, is more liable to metastasize than lung squamous carcinoma at the early stage.3,4 Therefore, exploring the molecular mechanisms of lung adenocarcinoma metastasis is essential, which could discover new therapeutic targets for treating lung cancer at an early stage.
Accumulating evidence suggests that protein arginine methylation is one of the most common post-translational modifications, which control kinase activity, protein stability, interaction, translocation, and localization. 5 Protein arginine methyltransferase 5 (PRMT5) belongs to the type II arginine methyltransferase that produces the symmetrical dimethylation of histone or non-histone protein substrates at the arginine residues, which are engaged in various fundamental cellular processes, including DNA replication, reparation and transcription, RNA splicing, protein degradation, cell metabolism, cell cycle progression, proliferation, ribosome biogenesis, anti-apoptosis, and signaling transduction.6–9 Dysfunction of PRMT5 is closely linked to different human cancers, including lung cancer. It has been reported that PRMT5 governs lung cancer cell growth and duplication through PI3K/Akt signaling cascades, whereas down-regulation of PRMT5 expression facilitates lung cancer cell apoptosis induced by resveratrol via Akt/glycogen synthase kinase 3β signaling pathway. 10 Previous studies have shown that PRMT5 was involved in tumorigenesis and inhibited tumor suppressor F-box and WD repeat domain-containing 7 to promote c-Myc expression and induce pancreatic cancer cell proliferation. 11 However, to date, few studies have revealed the biological functions of PRMT5 in the regulation of metastasis in human lung cancer, and the related molecular mechanisms are still unknown.
In this study, we aim to investigate the function of the PRMT5 and fibroblast growth factor receptor 3 (FGFR3)/Akt signaling axis in the epithelial-mesenchymal transition (EMT) of human lung cancer and uncover the crucial role of PRMT5/FGFR3/Akt in metastasis in human lung cancer and reveal the underlying molecular mechanisms.
Materials and Methods
Lung Cancer Tissues Collection
Fresh human lung cancer tissues and matched adjacent normal tissues were collected between January 2018 and January 2020 from patients who accepted surgical treatment in Minhang hospital, Fudan University. Total 15 matched tissue samples including, normal lung tissues, lung tumor and lymph nodes without metastasis and lymph nodes with metastasis, were collected and were immediately flash frozen in liquid nitrogen. Then, those samples were used for further experiments and assays. The study was approved by the Ethical Committee of Shanghai Jinshan Tinglin Hospital. The ethic approved number is SBKT-2017-12081.
Cell Culture and Chemicals
The ASTC-a-1 cells were obtained from the Department of Medicine, Jinan University (Guangzhou, China) and the A549 cells were obtained from ATCC. ASTC-a-1 and A549 cells were cultured in Dulbecco's Modified Eagle's Medium (1:1) (DMEM, Gibco, Thermo Fisher Scientific) supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma cat# F2442), 50 units/mL penicillin, and 50 mg/mL streptomycin. H1299 cells were cultured in RPMI-1640 containing with 10% (v/v) FBS, 50 units/mL penicillin, and 50 mg/mL streptomycin. The cells were maintained at 37 °C with 5% CO2 in an incubator. PRMT5 specific inhibitor GSK591 was purchased from Sigma (cat# SML-1751). The PRMT5 potent inhibitors GSK3326595 (cat# S8664) and EPZ015666 (cat# S7748) were purchased from Selleck Chemicals.
Plasmids
We generated the lentivirus containing 2 human PRMT5-shRNAs to knock down PRMT5 expression in lung cancer cells. The human PRMT5 targeting sequences were shown below: shRNA1, 5′-GGATAAAGCTGTATGCTGT-3′; shRNA2: 5′-GCCATCTATAAATGTCTGCTA-3′. In order to package the lentivirus containing PRMT5-shRNAs, the helper plasmids MD2G and PAX2 were used (Addgene). In addition, we generated the human Flag-PRMT5 plasmid to perform the gain-of-function experiments. The human PRMT5 cDNA was subcloned into the Flag vector and verified by sequencing.
Gene Expression Assay
In order to carry out gene expression analysis, the total RNA was extracted from human normal and lung cancer tissues or A549 cells using TRIzol reagent (Cat# 15596-018; Invitrogen) according to the manufacturer's protocol. Subsequently, an equal amount of RNA (1 µg) was used to perform the reverse transcription using Bio-Red PCR thermal cycler (C1000). Next, the SYBR green fluorescent Dye (cat# 1725272; Bio-Rad) was subjected to the quantitative real-time PCR (qRT-PCR) for gene expression analysis using an ABI7500 PCR machine (Applied BiosystemsTM). The following primers were used in this study: human PRMT5 forward: 5′-CCTGTGGAGGTGAACACAGT-3′ and revise: 5′-AGAGGATGGGAAACCATGAG-3′; FGFR3 forward: 5′-CATCCGGCAGACGTACACGC-3′ and revise: 5′-ACTGTACACCTTGCAGTGGA-3′; β-catenin forward: 5′-AAAGCGGCTGTTAGTCACTGG-3′ and revise: 5′-CGAGTCATTGCATACTGTCCAT-3′; Collagen I forward: 5′-GAGGGCCAAGACGAAGACATC-3′ and revise: 5′CAGATCACGTCATCGCACAAC-3; vimentin forward: 5′-AGTCCACTGAGTACCGGAGAC-3′ and revise: 5′-CATTTCACGCATCTGGCGTTC-3′; E-cadherin forward: 5′-ATTTTTCCCTCGACACCCGAT-3′ and revise: 5′-TCCCAGGCGTAGACCAAGA-3′; GAPDH, forward: 5′-CCATGTTCGTCATGGGTGTG-3′ and revise: 5′-CAGGGGTGCTAAGCAGTTGG-3′. GAPGH served as an internal control. The relative mRNA expression level was calculated by the method of ΔΔ-Ct.
Construction of PRMT5 Stable Knockdown Cell Line
To generate a PRMT5 stable depletion cell line, the lentivirus containing scramble or PRMT5-shRNAs was co-transfected along with the helper plasmids MD2G and PAX2 into 293 T cells using LipofectamineTM 3000 (cat# L3000015, Invitrogen) transfection reagent (Invitrogen, cat# 11668019) according to the manufacturer's protocol. The culture media was harvested, and the viral titer was pre-determined after 48 h. To further generate the PRMT5 stable knockdown cell line, A549 cells were infected with the indicated lentivirus with an equal amount of virus particles and then selected with puromycin (1 mg/mL, Sigma, cat# p9620) for 48 h. The non-infected cells were killed, and the PRMT5 depletion stable cells were used for the indicated experiments.
Cell Transfection
A549 cells were seeded into the 6-well plates and were transfected with flag-vector or human flag-PRMT5 plasmid when the cells were grown at around 70% using the LipofectamineTM 3000 (cat# L3000015, Invitrogen) transfection reagent according to the manufacturer's protocol. The cells were harvested for Western blotting analysis after 48-h post-transfection.
Western Blotting Analysis
Western blotting was performed as described previously. 27 Briefly, the total proteins were extracted from A549 cells or normal human tissues, and lung cancer tissues using the lysis buffer (20 mmol/L Tris, PH 7.4, 2 mmol/L EDTA, 2 mmol/L EGTA, 1 mmol/L sodium orthovanadate, 1% Triton X-100, 150 mmol/L NaCl, 50 mmol/L sodium fluoride, 0.1% SDS, and 100 mmol/L phenylmethylsulfonyl fluoride) and then were centrifuged for 10 min at 4 °C. The proteins were separated in sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE) and transferred to the PVDF membranes (cat#1620177; BioRed). Next, the membranes were washed 4 times with TBST for 10 min and blocked with 5% non-fat milk for 1 h at room temperature. The membrane was incubated with the following antibodies: PRMT5 (cat# sc-376937; Santa Cruz Biotechnology), Symmetric Di-Methyl Arginine Motif [sdme-RG] MultiMab™ (cat# 13222; Cell Signaling Technology), phospho-Ser473-Akt (cat# 4060; Cell Signaling Technology), total Akt (cat# 4691; Cell Signaling Technology), β-catenin (cat# 8480; Cell Signaling Technology), Collagen I (cat# ab34710; Abcam), FGFR3 (cat# 4574; Cell Signaling Technology), vimentin (cat# 5741; Cell Signaling Technology), E-cadherin (cat# 14472; Cell Signaling Technology), and β-actin (cat# sc-47778, Santa Cruz Biotechnology). After incubation, the membranes were washed 4 times with TBST for 5 min and incubated with goat anti-mouse conjugated to HRP secondary antibody (cat# sc-2005; Santa Cruz Biotechnology) or goat anti-rabbit conjugated to HRP secondary antibody (cat# sc-2004; Santa Cruz Biotechnology) for 2 h at room temperature. Immunoreactivity was detected by SuperSignal West Pico Chemiluminescent Substrate Western blotting detection reagents (cat# 34580; Thermo Fisher Scientific).
Immunofluorescence
A549 were cultured on glass coverslips and incubated overnight to establish adherence. The cells were fixed with 3.7%-4% paraformaldehyde for 15 min at room temperature and then permeabilized with ice-cold methanol for 15 min at −20 °C. The cells were incubated in blocking buffer (PBS containing 5% normal goat serum and 0.3% Triton X-100) for 1 h at room temperature, followed by incubation with anti-PRMT5 (cat# sc-376937; Santa Cruz Biotechnology), anti-proliferating cell nuclear antigen (cat# 13110; Cell Signaling Technology), anti-phospho-Ser473-Akt (cat# 4060; Cell Signaling Technology), and anti-actin antibody (cat# 4970; Cell Signaling Technology or cat# sc-47778; Santa Cruz Biotechnology) (diluted 1:100 in blocking buffer) at 4 °C overnight. Cells were washed 4 times for 5 min in PBS and then incubated for 2 h with Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (cat no. A-11034; Thermo Fisher) for p-ser473-Akt, PCNA, and actin; Alexa Fluor 568-conjugated goat anti-mouse secondary antibody (cat no. A-11004; Thermo Fisher) for PRMT5 (diluted 1:500 in blocking buffer) at room temperature. Nuclei were stained with DAPI (cat no. D9542; Sigma) for 15 min at room temperature before observation. The images were captured with a confocal microscopy system (LSM700, Zeiss).
Cell Proliferation Assay
A549, H1299, and ASTC-a-1 cells were seeded in 96-well plates and then maintained in the presence of vehicle or indicated PRMT5 inhibitors before adding 20 μL CellTiter 96 AQueous One Solution per well (Promega Corporation, Madison, WI. cat# G3582). Plates were incubated for 2 h, and then the OD490 (absorbance) was determined with a microplate reader (Tecan Infinite 200). Wells containing medium only served as background for the measurement. In some experiments, PRMT5 stable depletion A549 cells were seeded in 96-well plates and cultured at different time points, and the cell proliferation was measured as described above.
Statistical Analysis
All experiments were performed in triplicate under identical conditions, and the data were shown as means ± SD. Unpaired 2-tailed Student's t-test analyzed differences between the 2 groups. The difference with P < .05 was considered statistically significant.
Results
PRMT5/FGFR3/Akt Signaling Axis is Hyperactivated in Human Lung Cancer Tissues
It has been shown that PRMT5 is involved in various types of human cancer progression and is considered as an oncogene, 7 whereas how PRMT5 exerts its function in human lung cancer is still unclear. Our previous studies have shown that PRMT5 facilitated lung cancer cell growth and replication by activating Akt. 12 Nevertheless, how PRMT5 regulates Akt activity is still unknown. To validate the clinical relevance of PRMT5 expression in human lung cancer, we first analyzed the mRNA expression level of PRMT5 and FGFR3 using clinical samples. We found that PRMT5 and FGFR3 mRNA expression level was higher in lung cancer tissues than in normal tissues (Figure 1A). To further confirm these results, PRMT5 and FGFR3 protein expression was detected by Western blotting. As shown in Figure 1B, PRMT5 and FGFR3 protein expression was significantly increased in lung cancer tissues compared with normal tissues. Moreover, Akt phosphorylation was dramatically increased in lung cancer tissues, whereas the total Akt expression was unchanged. These results imply that PRMT5 regulates Akt activity by regulating FGFR3. Subsequently, we measured the PRMT5 and FGFR3 expression in normal tissues, lung cancer tissues, and with or without lymph node metastasis tissues. We found that PRMT5 and FGFR3 were overexpressed in lymph nodes with metastasis compared with normal tissues but not in lymph without metastasis (Figure 1C and D). Our results suggest that PRMT5/FGFR3/Akt signaling is closely related to lung cancer metastasis.

PRMT5/FGFR3/Akt axis is highly expressed in human lung cancer tissues. (A) The relative mRNA expression levels of PRMT5 and FGFR3 were measured by qRT-PCR in human lung cancer tissues and adjacent normal tissues (n = 15). **P < .01 versus normal tissues. (B) PRMT5, FGFR3, and phospho-Akt protein expression levels were detected by Western blotting in adjacent normal tissues (n) and human lung cancer tissues (t). (C and D) PRMT5 and FGFR3 expression levels were determined by Western blotting in adjacent normal tissues (n), human lung cancer tissues (t), and with (+) or without (−) lymph node metastasis.
Down-Regulation of PRMT5 Reduces FGFR3/Akt Signaling in Lung Cancer Cells
To investigate the effects of PRMT5 on FGFR3/Akt signaling and cell proliferation in lung cancer cells, we first generated the PRMT5 stable knockdown cell line (A549 cells). As shown in Figure 2A and B, silencing PRMT5 markedly reduced the expression levels of FGFR3 and Akt phosphorylation. Furthermore, down-regulation of PRMT5 also significantly decreased the cellular symmetric dimethylarginine (SDMA) expression, representing readout of PRMT5 enzymatic activity, suggesting that PRMT5 regulates FGFR3/Akt signaling in lung cancer cells. To further explore the effects of PRMT5 on FGFR3/Akt signaling and cell proliferation, immunofluorescence was carried out in PRMT5 depletion cells. As shown in Figure 2C, PRMT5 was located in the cytosol and cell surface (indicated by arrows) in control cells, implying that PRMT5 may directly interact with FGFR3, while PRMT5 expression almost completely disappeared in PRMT5 depletion cells, including cell surface. As shown in Figure 2D, phosphorylated Akt expression is visible, and activated Akt is located on the cell surface in control cells, while phosphorylated Akt expression almost completely disappeared in PRMT5 depletion cells, including cell surface (indicated by arrows). These results suggest that PRMT5 regulates FGFR3/Akt signaling in lung cancer cells. Next, the effects of PRMT5 depletion on cell proliferation in lung cancer cells were evaluated. As shown in Figure 2E, PCNA, a cell proliferation marker, was distinctly reduced in PRMT5 knockdown cells compared with control cells, and the cell viability was dramatically decreased as well (Figure 2F). Altogether, our results demonstrate that PRMT5 controls lung cancer cell proliferation via FGFR3/Akt signaling.

Silencing of PRMT5 impairs FGFR3/Akt signaling in lung cancer cells. (A) A549 cells were infected with lentivirus containing PRMT5-shRNAs, and the indicated protein expression levels were detected by Western blotting. Representative pictures were shown. (B) The indicated protein expression levels were quantified. *P < .05 versus Scr, n = 3. (C) The PRMT5 expression and localization were measured by immunofluorescence in PRMT5 depletion A549 cells. Representative pictures were shown (n = 3). Scale bar = 50 µm. (D) The phospho-Akt-Ser473 expression and localization were measured by immunofluorescence in PRMT5 depletion A549 cells. Representative pictures were shown (n = 3). Scale bar = 50 µm. (E) The proliferation marker PCNA expression was measured by immunofluorescence in PRMT5 depletion A549 cells (n = 3). Scale bar = 50 µm. (F) Cell viability was determined by MTS assay in PRMT5 depletion A549 cells (n = 4). *P < .05 versus Scr, n = 3.
Inhibition of PRMT5 Represses FGFR3/Akt Signaling in Lung Cancer Cells
To further confirm our hypothesis above, we block PRMT5 activity with 3 specific inhibitors (EPZ, GSK595, and GSK591) which are commonly used in many investigations and clinical trial studies. As shown in Figure 3A, inhibiting PRMT5 activity by specific inhibitors markedly impaired the expression levels of FGFR3 and Akt phosphorylation. Moreover, inhibition of PRMT5 also significantly decreased the cellular SDMA expression. To further evaluate the effects of PRMT5 inhibitors on FGFR3/Akt signaling and cell proliferation, immunofluorescence was performed upon treatment of PRMT5 inhibitor GSK591. As shown in Figure 3B, phosphorylated Akt expression is visible and activated Akt located on the cell surface in cells treated with vehicle, while phosphorylated Akt expression was almost completely disappeared in cells treated with PRMT5 inhibitor GSK591, including cell surface. As shown in Figure 3C and D, PCNA, a cell proliferation marker, was distinctly reduced in cells treated with PRMT5 inhibitor compared with control cells treated with vehicle. In addition, the cell viability was dramatically decreased as well under PRMT5 inhibitor treatment compared with control cells (Figure 3E). Collectively, these results confirm our findings that PRMT5 regulates lung cancer cell proliferation via FGFR3/Akt signaling.
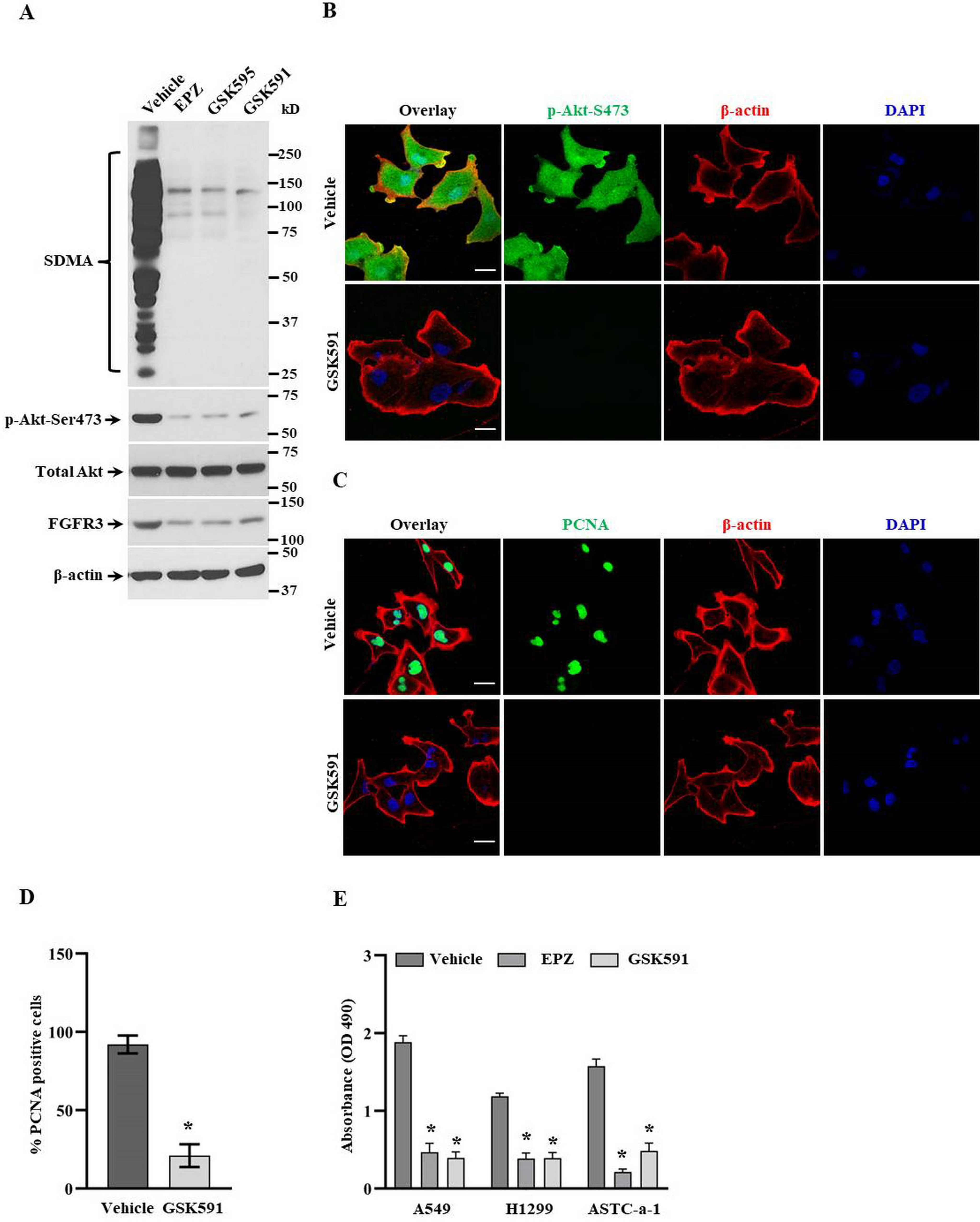
Inhibiting PRMT5 suppresses FGFR3/Akt signaling in lung cancer cells. (A) A549 cells were treated with indicated PRMT5 inhibitors, and the related protein expression levels were detected by Western blotting. Representative pictures were shown. (B) The phospho-Akt-Ser473 expression and localization were measured by immunofluorescence upon GSK591 treatment. Representative pictures were shown (n = 3). Scale bar = 50 µm. (C) The proliferation marker PCNA expression was measured by immunofluorescence upon GSK591 treatment. Representative pictures were shown (n = 3). Scale bar = 50 µm. (D) PCNA-positive cells were quantified upon GSK591 treatment. One hundred cells were counted for each group. *P < .05 versus Vehicle. (E) Cell viability was determined by MTS assay upon GSK591 treatment (n = 4). *P < .05 versus Vehicle.
PRMT5 Regulates Lung Cancer Cell Metastasis Through FGFR3/Akt Signaling Axis
We next determined EMT biomarkers expression to probe the molecular basis for PRMT5-mediated cell metastasis, including vimentin, collagen I, β-catenin, and E-cadherin. As shown in Figure 4A and C, down-regulation of PRMT5 significantly reduced the expression of mesenchymal markers, including vimentin, collagen I, and β-catenin, both at mRNA and protein level, whereas PRMT5 depletion dramatically induced epithelial marker E-cadherin expression. In order to further confirm our results, we reintroduced Flag-PRMT5 in PRMT5 depletion cells. As shown in Figure 4B and D, the re-expression of PRMT5 significantly increased the expression of mesenchymal markers, including vimentin, collagen I, and β-catenin, both at mRNA and protein level, whereas PRMT5 re-expression dramatically reduced epithelial marker E-cadherin expression. Finally, we also found that re-expression of PRMT5 markedly elevated the expression levels of FGFR3 and Akt phosphorylation in PRTM5 depletion cells, which are closely related to tumor cell metastasis. Altogether, our findings suggest that PRMT5 modulates lung cancer cell metastasis by activating FGFR3/Akt signaling axis.

PRMT5/FGFR3/Akt signaling axis controls lung cancer cell metastasis. (A) The relative mRNA expression levels of metastasis markers were measured by qRT-PCR in PRMT5 depletion A549 cells (n = 4). *P < .05 versus Scr. (B) The relative mRNA expression levels of metastasis markers were measured by qRT-PCR in PRMT5 depletion A549 cells, which overexpressed Flag-PRMT5 (n = 4). *P < .05 versus Vector. (C). The indicated protein expression levels were detected by Western blotting in PRMT5 depletion A549 cells. Representative pictures were shown (n = 3). (D) The indicated protein expression levels were detected by Western blotting in PRMT5 depletion A549 cells, which overexpressed Flag-PRMT5. Representative pictures were shown (n = 3).
Discussion
So far, it is well known that PRMT5 is considered as an oncoprotein and engaged in various human cancer progression and development by regulating different signaling pathways. 13 Thus, PRMT5 has become an up-and-coming candidate for treating human cancers. Nevertheless, it is still completely unknown whether PRMT5 controls cancer cell metastasis, and the related molecular mechanisms are also obscure, especially in human lung cancer. The findings in the current study showed that PRMT5/FGFR3 and phosphorylated Akt were highly expressed in human lung cancer tissues and were closely related to lymph nodes with metastasis (Figure 1). This phenomenon implies that PRMT5 may control lung cancer metastasis by regulating FGFR3/Akt signaling axis. Our further investigation showed that silencing PRMT5 by lentivirus-mediated shRNAs or inhibiting PRMT5 by specific inhibitors, not only reduced the expression levels of FGFR3 and phosphorylated Akt but also prevented lung cancer cell proliferation (Figures 2 and 3). These results further confirmed our hypothesis that PRMT5 governed lung cancer cell growth by regulating FGFR3/Akt signaling axis. Moreover, down-regulation or re-expression of PRMT5 decreased or promoted EMT-related markers (Figure 4) and FGFR3/Akt signaling axis (Figures 2 to 4), including vimentin, collagen I, and β-catenin, respectively, indicating that PRMT5 modulates lung cancer cell metastasis activating FGFR3/Akt signaling axis. These results suggest that PRMT5 could be served as a novel target for treating human lung cancer therapy and raise the possibility that PRMT5 promotes human lung cancer cell proliferation and metastasis by controlling FGFR3/Akt signaling axis (Figure 5).

The proposed model of the PRMT5/FGFR3/Akt axis regulates metastasis in human lung cancer.
A previous study has demonstrated that down-regulation of PRMT5 repressed cell duplication at the G1 phase, and PRMT5 activity was regulated by CDK4/cyclin D1 complex through the phosphorylation of MEP50. 14 Moreover, PRMT5 promotes liver cancer cell proliferation by inhibiting BTG2 expression and up-regulation of cyclin E1 and cyclin D1. 15 Furthermore, PRMT5 methylated CDT1 and E2F1 to promote cells entry G1-to S-phase and cycle progression.14,16,17 These observations indicate that PRMT5 is essential for cell proliferation via regulation of cell cycle progression and imply that PRMT5 is a pivotal upstream mediator for cancer cell proliferation. In the present study, we showed that silencing or inhibiting PRMT5 by shRNAs or inhibitors prevented lung cancer cell growth, which not only suggested that PRMT5 regulates cancer cell proliferation and metastasis but also indicates that PRMT5 is a critical upstream regulator for human lung cancer cell growth.
Our previous study showed that PRMT5 facilitated lung cancer cell growth by regulating AKT activity. 12 However, how PRMT5 controls AKT activity is still unknown. More and more evidence has shown that FGFR3 was found in NSCLC, which has a crucial role in lung cancer initiation and progression. 18 Additionally, FGFR3 played a vital role in a variety of biological processes, including differentiation, development, cell metabolism, proliferation, invasion, angiogenesis, and carcinogenesis by several intracellular pathways, 19 including the PI3K/AKT 20 and Ras/Raf/MEK signaling pathways. 21 The current study showed that PRMT5 controlled lung cancer cell proliferation by regulating FGFR3/Akt signaling axis. Furthermore, a recent novel study reported that PRMT5 interacts with AKT and methylated AKT-R15 to promote tumor metastasis, providing direct evidence to show how PRMT5 controls AKT activity as well. 22
EMT is the most critical event in human cancer progression, development, and metastasis, which is also linked to the functions of various human cancers, including migration, invasion, and tumor initiation.23,24 It has been shown that EMT was tightly controlled by Akt in various types of human cancer, including lung cancer. Akt is the master regulator for tumor cell survival, proliferation, and anti-apoptosis. 25 Dysfunction of Akt signaling has participated in different human diseases, such as autoimmune, metabolic diseases, cardiovascular disorders, and cancer. 26 However, how PRMT5 regulates EMT in human lung cancer cells and the underlying molecular mechanism remains unknown, although previous studies have shown that PRMT5 regulates cancer cell proliferation by PI3K/PTEN/Akt signaling. In the present study, we showed that PRMT5 regulated EMT by activating FGFR3/Akt axis in lung cancer cells using PRMT5-stable depilation cells. A recent study showed that PRMT5 was positively associated with FGFR3 expression and promoted lung cancer growth and metastasis through miR-99. 28 Nevertheless, this study did not fully show how PRMT5 regulates EMT in human lung cancers; the related mechanism is still unclear. In the present study, we showed that PRMT5 and FGFR3 were highly expressed in human lung cancer tissues and were closely related to lymphatic metastasis. Moreover, down-regulation of PRMT5 by lentivirus-mediated shRNAs or inhibition of PRMT5 by specific inhibitors attenuates FGFR3 expression, Akt phosphorylation, and lung cancer cell proliferation. Further studies showed that silencing PRMT5 impairs EMT-related markers, including vimentin, collagen I, and β-catenin. Conversely, ectopic expression of PRMT5 increases FGFR3 expression, Akt phosphorylation, and EMT-related markers, suggesting that PRMT5 regulates metastasis probably through the FGFR3/Akt signaling axis.
There are limitations to this work. While PRMT5/FGFR3/Akt signaling axis control lung cancer cell proliferation and metastasis, future studies should further identify how PRMT5 directly or indirectly controls FGFR3 activity. In this work, the number of clinical tumor samples from human individuals was relatively small. Future investigation will be required to confirm the positive correlation among PRMT5, FGFR3, and p-Akt. Moreover, the mechanism by which PRMT5 regulates FGFR3 is still not fully understood, but it may also involve methylation of FGFR3 or other related targets, which was not directly detected in this work. Finally, as noted above, the downstream factors of the PRMT5/FGFR3 signaling axis in metastasis need to be further identified.
Conclusion
Our findings demonstrate that PRMT5 is an ectopic expression in human lung cancer tissues. In addition, our studies uncover that PRMT5 promotes human lung cancer cell proliferation probably via FGFR3/Akt signaling axis. The noteworthy feature of our findings is that PRMT5 regulates the activation of the FGFR3/Akt signaling axis, which then affects the expression of the downstream targets involved in metastasis. Most importantly, we established the relationship between PRMT5 and FGFR3/Akt signaling axis in human lung cancer. The new insights into regulating the FGFR3/Akt signaling axis governed by PRMT5 elicit a new mechanism of PRMT5 carcinogenic function in human lung cancer.
Footnotes
Abbreviations
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The study was approved by the Ethical Committee of Shanghai Jinshan Tinglin Hospital. The ethic approved number is SBKT-2017-12081.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Project of Shanghai Municipal Health Commission (201940102, 201940280), Specialist Fund of Central Hospital of Shanghai Minhang District (YJXK-2021-02-005), and Supported Specialist Fund of Shanghai Jinshan Tinglin Hospital (TLZK2020-F01).