
Abstract
Introduction
With an increasing incidence, hepatocellular carcinoma (HCC) is one of the leading causes of cancer-associated mortality. 1 Although therapeutic advances have been achieved in the treatment of HCC, the overall prognosis for HCC patients remains poor, since early stage of HCC usually lack typical clinical symptoms and 70%–80% of HCC patients are diagnosed at an advanced stage. 2 Therefore, a better understanding of underlying molecular mechanisms of HCC progression and identification of potential biomarkers will contribute to earlier diagnosis and effective treatment.
MicroRNAs (miRNAs) belong to a class of single-stranded non-cording RNAs consisting of 19-23 nucleotides. 3 The most prominent function of miRNAs is their negative effects on target gene expression either by mRNA degradation or translational repression, 4 through which they play critical roles in the regulation of numerous biological processes of eukaryotic cells.5,6 Therefore, dysregulation of miRNAs is correlated with the occurrence of various diseases including cancer.7,8 For instance, miR-25 and miR-93 mediate hypoxia-induced immunosuppression of breast cancer by repressing the activity of the NCOA3-cGAS pathway, 9 miR-26b-5p suppresses chemoresistance in breast cancer by targeting serglycin, 10 and abnormal expression of multiple miRNAs (miR-125b, miR-105, miR-548p, etc.) is also essential during the progression of HCC.11–13 MiR-1205, a miRNA originally identified in a genomically unstable region of human chromosome 8q24, 14 has been reported to be abnormally expressed in multiple tumors (prostate cancer, colorectal cancer, glioma, etc.), resulting in the inhibition or progression of these tumors.15–17 Recent findings have demonstrated that CircFN1 mediates sorafenib resistance of HCC cells by sponging miR-1205 and regulating E2F1 Expression, indicating a tumor-suppressing role of miR-1205 in HCC. 18 However, the expression patterns and underlying mechanisms of miR-1205 remain unclear yet.
Casein kinase 2 beta (CSNK2B) is a regulatory subunit of casein kinase II (CK2), which plays key roles in multiple biological functions such as cell proliferation, differentiation, apoptosis, and metabolism by phosphorylating the serine and threonine residues of specific proteins. 19 In recent years, CSNK2B has been implicated in the development of human malignancies. For example, CSNK2B contributes to the development of colorectal cancer by activating the mTOR signaling, 20 it also acts as an oncogene in gastric cancer and HCC.21,22 Despite these studies suggested a cancer promoting role of CSNK2B, the underlying mechanisms of the abnormal CSNK2B expression in HCC remain poorly understood.
In the present study, we resorted to explore the role of miR-1205 in HCC. Our findings demonstrated that miR-1205 is a potential tumor suppressor gene in HCC by directly targeting CSNK2B and inhibiting its expression at the transcriptional level, which finally suppressed HCC cell proliferation via inactivation of the CDK4/pRb cell cycle pathway. This study will provide new insights into the regulatory mechanisms of miR-1205 in HCC tumorigenesis.
Materials and Methods
Sample Collection
Twenty pairs of fresh HCC tumor tissues and corresponding adjacent non-tumor hepatic tissues were obtained from patients who were diagnosed with HCC based on histopathological examination of the resected tissues in Hwa Mei Hospital, University of Chinese Academy of Sciences. The fresh tissues were snap-frozen and stored in liquid nitrogen prior to RNA extraction. Ethics approval was granted by the Institutional Ethics Committee of Hwa Mei Hospital, University of Chinese Academy of Sciences, and informed consent was signed by all patients enrolled in the present study.
Cell Culture and Reagents
HCC cell lines PLC/PRF/5 (TCHu119), Hep3B (SCSP-5045), HepG2 (TCHu 72) were purchased from the Shanghai cell bank, Chinese Academy of Sciences, Shanghai, China. HCC-LM3 and Focus cells were taken from our laboratory stocks. All cells were cultured using Dulbecco modified Eagles medium (DMEM, Corning, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, CA, USA) and 1% penicillin/streptomycin (P/S, Corning, NY, USA) in a humidified incubator at 37°C with 5% CO2. Palbociclib (a highly selective CDK4/6 inhibitor) was purchased from Shanghai Selleck Chemicals (#S1116, Shanghai, China).
Cell Transfection
CSNK2B overexpression plasmids, small-inference RNAs against CSNK2B, overexpression lentivirus of miR-1205, miRNA inhibitor, miRNA mimics, and their controls used in this study were constructed and purchased from GenePharma Co., Ltd (Shanghai, China). Cell transfection was performed using the Lipofectamine® 3000 Transfection Reagent (Invitrogen, CA, USA) following the manufacturer's protocol. Lentivirus transduction was performed by supplementation of 4 μg/mL polybrene followed by puromycin selection (1.5 μg/mL, Sigma-Aldrich, MO, USA).
RNA Extraction and Quantitative Real-Time PCR (q-PCR) Analysis
Cellular and Tissue RNAs were isolated using Trizol reagent (Invitrogen, CA, USA) according to standard protocols. The reverse transcription reaction step was then performed with a PrimeScript reverse transcriptase kit (TaKaRa, Shiga, Japan). An ABI 7500 real-time quantitative PCR instrument (Applied Biosystems, CA, USA) was used to perform qPCR analysis with QuantiTect SYBR Green PCR Kit (Qiagen, Frankfurt, German) as per the manufacturer's specifications. RNU6B (U6) and GAPDH were used as internal control for miR-1205 and CSNK2B, respectively. Each experiment was performed in triplicate and repeated for at least 3 times independently. Primers used in the present study were listed as follows: miR-1205, F: 5ʹ-GCAGGGTTTGCTTTGAGTACTTCCTTCCTGTCA-3ʹ, R-5ʹ-GTCCAGTTTTTTTTTTTTTTTACAFACT5ʹ-. U6, F: 5ʹ-CGCTTCGGCAGCA CATAT-3ʹ, R: 5ʹ-AAATATGGAACGCTTCACGA-3ʹ. CSNK2B, F: 5ʹ-TGAGCAGGTCCCTCACTACC-3ʹ, R: 5ʹ-GTAGCGGGCGTGGATCAAT-3ʹ. GAPDH, F: 5ʹ-GGAGCGAGATCCCTCCAAAAT -3ʹ, R: 5ʹ-GGCTGTTGTCATACTTCTCATGG -3ʹ.
Western Blot Analysis
Total proteins were extracted using RIPA lysis buffer (Beyotime, Shanghai, China) supplemented with phosphatase and protease inhibitors (Roche Diagnostic, Mannheim, Germany) and protein concentration was quantified with a BCA Protein Assay Kit (Thermo Scientific, MA, USA) according to the manufacturers’ protocols. Equal amounts of protein samples (20 μg) were electrophoresed on a 10% gradient SDS-polyacrylamide gel (SDS-PAGE) and transferred onto a nitrocellulose membrane using the Trans-Blot Turbo transfer system (Bio-Rad, CA, USA). Incubation with primary antibodies was performed overnight at 4 °C, followed by washing with PBS–0.1% Tween 20 (PBST, Shenggong Biotech, Shanghai, China) for three times and incubation with fluorescent secondary antibodies for 1 h at room temperature. Protein bands were visualized using an Odyssey Sa Infrared Imaging System (LI-COR, NE, USA). GAPDH protein served as an internal reference. The primary antibodies used in the present study were as below: anti-CSNK2B (1:500, #20234-1-AP, ProteinTech, Wuhan, China), anti-GAPDH (1:50000, #60004-1-Ig, ProteinTech, Wuhan, China), CDK4 (1:500, #20234-1-AP, ProteinTech, Wuhan, China), (1:500, #20234-1-AP, ProteinTech, Wuhan, China), anti-CDK2 (1:500, #10122-1-AP, ProteinTech, Wuhan, China), CDK6 (1:500, #14052-1-AP, ProteinTech, Wuhan, China), anti-CCND1 (1:500, #26939-1-AP, ProteinTech, Wuhan, China), anti-CCNE1 (1:500, #11554-1-AP, ProteinTech, Wuhan, China), anti-Rb (1:1000, #9309, Cell Signal Technology, MA, USA) and anti-pRb (1:500, #8516, Cell Signal Technology, MA, USA).
Cell Proliferation Assay
Cell proliferation rates of HCC cell lines were measured using Cell Counting Kit-8 assays (CCK-8, Dojindo, Kumamoto, Japan). Briefly, the indicated cells were seeded into a 96-well plate with a density of 3 × 103 cells per well and cultured under normal conditions. At each time point (1, 2, 3, 4, 5 d), 10 µL of CCK-8 solution was added into each well. After incubation for 1 h at 37°C, the absorbance at 450 nm was determined using a Spectra Max M5 (Molecular Devices company, CA, USA). Growth curves were constructed to compare cell proliferation rates between two groups. All experiments were conducted in triplicate and repeated at least for three times independently.
Colony Formation Assay
Colony forming ability of HCC cells was evaluated using colony formation assays. The indicated cells were seeded into a six-well plate at a density of 1 × 103 cells/well. After culture for 1 week, the cells were fixed with 4% paraformaldehyde at room temperature for 10 min and stained with 0.1% crystal violet (#C3886, Sigma-Aldrich, MO, USA) at room temperature for 15 min. Colonies were photographed and counted to assess the colony forming ability of HCC cells. All experiments were done in triplicate and repeated for more than 3 times.
Online miRNA Target Prediction, Gene Expression and Survival Analysis
To predict potential target genes of miR-1205, miRDB (http://mirdb.org/miRDB/) and TargetScan (http://www.targetscan.org) were used. CSNK2B expression from The Cancer Genome Atlas database (TCGA, https://tcga-data.nci.nih.gov/tcga/) was analyzed using GEPIA (http://gepia.cancer-pku.cn), a bioinformatics tool for analyzing and visualizing TCGA. Kaplan–Meier survival analysis was carried out using publicly available miRNA expression datasets (miRpower for pan-cancer) from Kaplan–Meier (KM) plotter (https://www.kmplot.com).
Dual Luciferase Reporter Assay
Luciferase reporter plasmids either wild type (WT) or mutated (MUT) 3ʹUTR sequence of CSNK2B were constructed and purchased from Hanyin, Shanghai, China. By site-directed mutagenesis, mutant constructs in KLF4 binding sequence CCUGCAG to GGGGCAG were generated. Dual-luciferase reporter assays were performed in 24-well plates. The cells were co-transfected with pGL3-CSNK2B-WT or pGL3-CSNK2B-MUT firefly luciferase reporter plasmids along with pRL-SV40 renilla luciferase construct. After 24 h, the luciferase activities were tested using the Dual-Luciferase Reporter Assay System (Promega, WI, USA) according to the protocol of manufacturer. Renilla luciferase was used as the internal control.
Animal Experiments
The current study conformed with the ARRIVE 2.0 guidelines. 23 Four to six weeks old male nude mice (BALB/c) were purchased from Slac Laboratories (Shanghai, China). To establish a subcutaneous xenograft model, 2 × 106 HCC-LM3 cells with stable overexpression of miR-1205 or the control cells were randomly inoculated into the left armpit region of nude mice, then the mice were raised for 4 weeks under normal feeding conditions (constant temperature of 25 °C, relative humidity of 50 ± 10%, alternating light of 12 h, the activity of cage feeding in mice was not limited, and the diet could be freely used). Tumor volume was recorded every week. After 4 weeks, mice were euthanized, tumors were resected, weighed, and photographed.
Cignal Finder 10-Pathway Reporter Array
Cignal Finder 10-Pathway Reporter Array (Qiagen, Hilden, Germany) was performed in accordance with the manufacturer's protocols. In brief, we seeded suspended cells into 96-well plates that contained luciferase reporters to target common cancer pathway, then cells were incubated in a 5% CO2 incubator for 24 h for the detection of luciferase activity using the Dual-Luciferase Reporter Kit (Promega, Wisconsin, USA).
Statistical Analysis
All statistical analyses were performed with GraphPad Prism v8.0 (Graphpad Software Inc). Quantitative data were presented as mean ± SD. Unpaired Student's t-test (two-tailed) and Mann–Whitney test were performed between two groups and one-way analysis of variance (ANOVA) followed by Bonferroni's multiple comparison tests were used for statistical comparison between three or more groups. A value of P < 0.05 was considered statistically significant (*P < .05; **P < .01).
Results
MiR-1205 Inhibits HCC Cell Proliferation
in Vitro
and
in Vivo
In several HCC cell lines, miR-1205 expression was examined. The results showed that expression levels of miR-1205 were roughly identical in the cell lines used (Figure S1). To investigate the role of miR-1205 in HCC cell growth, human miR-1205 inhibitor or scrambled-miRNA inhibitor (control) was transiently transfected into Hep3B and HepG2 cells, respectively. Further qPCR analysis confirmed that miR-1205 level in HCC cells was substantially decreased by miR-1205 inhibitor compared to the control group (Figure 1A). Then CCK-8 cell proliferation assays were conducted after silencing of miR-1205 expression in Hep3B and HepG2 cells. As shown in Figure 1B, decreased miR-1205 expression significantly facilitated HCC cell growth comparing with the control group. Besides, miR-1205 expression in focus and HCC-LM3 cells was increased by transfection with miR-1205 mimics (Figure 1C). In contrast, cell growth rates in cells with miR-1205 overexpression were remarkably lower than the control cells (Figure 1D). A lentivirus expression system with puromycin resistance gene was used to increase miR-1205 expression stably in HCC cells. Colony formation assays also showed that miR1205 overexpression significantly decreased the colony number formed from Focus and HCC-LM3 cells (Figure 1E). In addition, similar effects in tumor growth were observed in a xenograft model of nude mice, indicating that increased miR-1205 expression was associated with attenuated tumorigenesis of HCC in vivo (Figure 1F). Taken together, these observations demonstrated that miR-1205 acts as a suppressive role in the regulation of HCC cell proliferation in vitro and in vivo.

MiR-1205 inhibits HCC cell proliferation in vitro and in vivo. (A) and (C) MiR-1205 inhibitor or mimics was transfected into Hep3B, HepG2, Focus, or HCC-LM3, respectively, and miR-1205 levels in different groups were detected by qPCR analysis. (B) and (D) After transfection with miR-1205 inhibitor or mimics, CCK-8 cell proliferation assays were performed to measure cell viability. (E) The effects of miR-1205 overexpression or inhibition on colony formation ability of Focus and HCC-LM3 cells were measured by colony formation assays. (F) Xenograft images (left) and analyses of tumor weight (middle) and volume (right) were shown. *P < .05, **P < .01.
MiR-1205 Directly Targets CSNK2B Gene
Since the main function of miRNAs is post-transcriptional regulation of their target gene expression, we next searched for putative miR-1205 target genes using online miRNA target prediction databases (TargetScan, miRDB, and miRWalk). Among the 31 overlapping target genes in these three databases, CSNK2B, known to be significantly associated with the progression of various cancer types including HCC, drew our attention (Figure 2A), and the binding site of the putative targeted gene and the mutated site of miR-1205 on the 3ʹUTR region of CSNK2B were shown in Figure 2B. Next, we verified whether miR-1205 regulates the expression of CSNK2B in HCC by qPCR analysis. As exhibited in Figure 2C, overexpression of miR-1205 significantly suppressed CSNK2B mRNA level in Focus and HCC-LM3 cells, whereas silenced miR-1205 expression resulted in opposite effects. Consistently, CSNK2B protein level in HCC cells was inhibited by miR-1205 (Figure 2D and Figure S2), indicating that miR-1205 negatively regulates CSKN2B expression in HCC cells. Moreover, the dual luciferase reporter assays successfully validated their interaction, as evidenced by decreased activity of wild-type (WT) but not the mutated (MUT) CSNK2B 3ʹUTR reporter (Figure 2E). Collectively, the above findings indicated that miR-1205 directly targets CSNK2B and inhibits its transcription in HCC cells.

CSNK2B is a target gene of miR-1205. (A) The overlapping target genes were predicted using different online tools (TargetScanHuman, miRWalk, and miRDB). (B) An illustration of the predicted and mutated miR-1205 binding site in the reporter assay construct. CSNK2B mRNA (C) and protein (D) levels in HCC cell lines transfected with miR-1205 inhibitor or mimics were assessed by qPCR analysis. (E) Bar graph showing the difference in normalized luciferase activities of wild type (WT) or mutant (MUT) CSNK2B 3ʹ-UTR in HCC cells transduced with miR-1205 lentivirus (LV-miR-1205) and a control lentivirus (LV-miR-Con). RLU, relative luciferase units; *P < .05, **P < .01.
CSNK2B Mediates the Effects of miR-1205 on HCC Proliferation
Previous studies have reported that CSNK2B can act as an oncogene in human malignancies including HCC,20,21 we thus investigate whether miR-1205 inhibits HCC proliferation by regulating CSNK2B expression. Specific siRNAs targeting CSNK2B (siCSNK2B-1/2) or CSNK2B-expressing plasmids were transfected into HCC cells (Figure 3A and Figure S3), and CCK-8 cell proliferation assays were performed. As shown in Figure 3B, overexpression of CSNK2B significantly promoted PLC/PRF/5 cell proliferation ability, whereas Focus cells with silenced CSNK2B expression showed slower growth rates as compared to their control cells, respectively. Furthermore, stable HCC-LM3 and PLC/PRF/5 cell lines overexpressing CSNK2B were generated via lentivirus, and colony formation assays demonstrated that overexpression of CSNK2B remarkably increased the colony formation ability of HCC cells (Figure 3C), which is consistent with previous studies. More importantly, the effects of miR-1205 overexpression or inhibition on HCC proliferation were almost counteracted by enforced or silenced expression of CSNK2B, respectively (Figure 3D-E). Therefore, the above-described observations provide evidence that the effects of miR-1205 on HCC proliferation were mediated by CSNK2B.

MiR-1205 inhibits HCC cell proliferation by regulating CSNK2B expression. (A) Representative western blot images showing the efficiencies of CSNK2B RNAi depletion in Focus (left) or overexpression plasmid in PLC/PRF/5 (right). (B) Growth curves of HCC cells transfected with CSNK2B-expressing plasmid (left) or CSNK2B-specific siRNAs (right) were determined using CCK-8 kit for 5 days, respectively. (C) Colony formation assays showing the effects of CSNK2B on colony formation ability of HCC cells. Representative images (left) and quantitative analysis on colony numbers (right) were shown. (D) and (E) Colony formation assays were performed to evaluate the effects of altered CSNK2B expression on the function of miR-1205. Quantitative analysis of colony numbers was shown. *P < .05, **P < .01.
CDK4/p-Rb/E2F Pathway is a Downstream Effector of CSNK2B
To explore potential mechanisms involved in the function of CSNK2B in HCC, a Cignal Finder 10-Pathway Reporter Array was conducted. Intriguingly, among the cancer-associated pathways, cell cycle/pRb-E2F pathway was significantly activated by CSNK2B overexpression (Figure 4A). Considering that cell cycle is regulated by distinct cyclin-dependent kinases (CDKs) and specific cyclin partners at phase transition, 24 we next investigate whether CSNK2B regulate expression of these proteins. As shown in Figure 4B and Figure S4A, CSNK2B significantly enhanced CDK4 expression in PLC/PRF/5 cells, while no significant changes of CDK2, CDK6, CCND1, and CCNE1 levels were observed between two groups, hinting us that CSNK2B possibly activates cell cycle pathway by regulating CDK4 expression. Consistently, CDK4 and its’ downstream signaling protein-phosphorylated retinoblastoma protein (p-Rb) levels were strongly inhibited by knockdown of CSNK2B in Focus cells, while total Rb level showed no significant change (Figure 4C and Figure S4B), suggesting that CSNK2B can activate the CDK4/p-Rb cell cycle pathway. To investigate whether CDK4 mediated the effects of CSNK2B on HCC cell proliferation, a Food and Drug Administration (FDA)-approved CDK4/6 inhibitor Palbociclib was used to inhibit CDK4/p-Rb pathway activity, and colony formation assays indicated that suppression of CDK4 remarkably attenuated the promotion of CSNK2B overexpression on HCC cell proliferation (Figure 4D). These findings suggested that CDK4/p-Rb/E2F pathway mediates the biological function of CSNK2B in the promotion of HCC cell proliferation.

CSNK2B activated CDK4/p-Rb/E2F cell cycle pathway. (A) Cignal Finder 10-Pathway Reporter Array data in HCC-LM3 cells upon CSNK2B overexpression. (B) Representative western blot images showing the effects of CSNK2B overexpression on key protein expression of cell cycle pathway. (C) Western blot analysis showing an inhibition of CDK4 expression by CSNK2B knockdown. (D) Colony formation assays were performed to evaluate whether inhibition of CDK4/p-Rb/E2F cell cycle pathway weakens the effects of CSNK2B overexpression on HCC cell proliferation. Representative images (upper) and quantitative analysis on colony numbers (bottom) were shown. *P < .05, **P < .01.
MiR-1205 Expression is Reduced in HCC Tissues and Negatively Associated with CSNK2B Expression
We analyzed TCGA-HCC RNA-seq data by online tools and found that CSNK2B mRNA expression was higher in HCC tumor tissues than in normal tissues (Figure 5A) and gradually increased with the progress of tumor stages (Figure 5B), which is in line with previous studies. Further Kaplan–Meier survival analysis showed that higher CSNK2B level was correlated with worse prognosis of patients with HCC (Figure 5C). To search potential association between miR-1205 and CSNK2B in HCC, qPCR analysis was performed to detect their expression in 20 paired HCC tumor and adjacent non-cancerous tissues. As expected, miR-1205 level in HCC tissues was significantly lower than in adjacent non-cancerous tissues (P < .01, Figure 5D), whereas higher CSNK2B expression was observed in HCC tissues (P < .01, Figure 5E). Furthermore, miR-1205 and CSNK2B levels in HCC were negatively correlated with each other (r = −0.3585, P < .01, Figure 5F), which was consistent with our experimental results. Taken together, the above results provided substantial evidence that miR-1205 functions as a tumor suppressor gene by inhibiting CSNK2B expression in HCC.
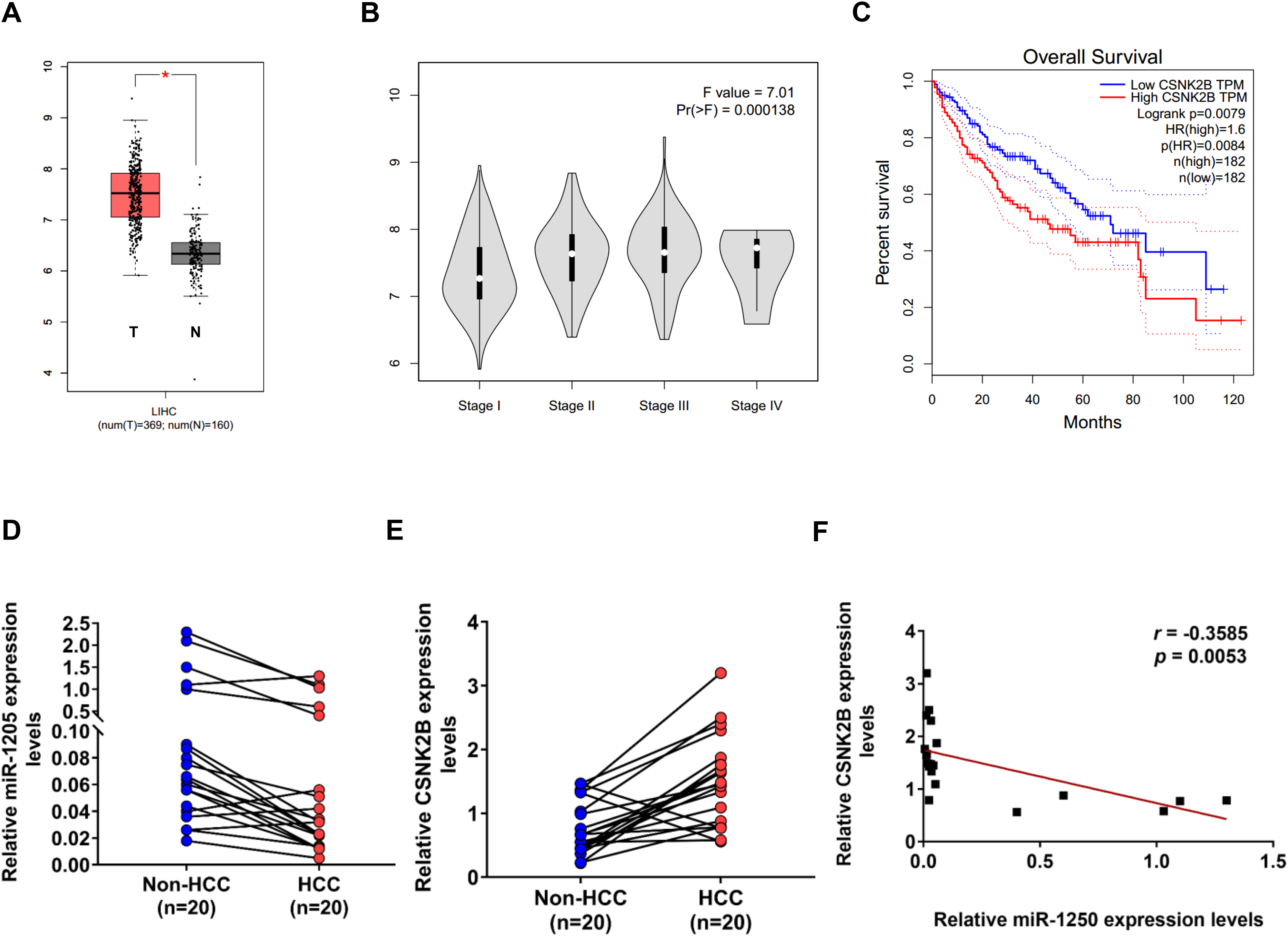
Decreased miR-1205 expression is negatively correlated with increased CSNK2B expression in HCC tissues. CSNK2B expression in HCC tissues (A) and its correlation with tumor stage (B) was analyzed using Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/) based on data from The Cancer Genome Atlas (TCGA). (C) The relevance between CSNK2B expression and overall survival rate of HCC patients was assessed by GEPIA (P = .0084). (D) and (E) QPCR analysis of miR-1205 D and CSNK2B E mRNA levels in 20 paired human HCC tissues and non-cancerous liver tissues (non-HCC). (F) Pearson correlation analysis showing a negative correlation between miR-1205 and CSNK2B expression in HCC (r = −0.3585, P < .01).
Discussion
Although the advances in precision medicine and targeted therapy have improved the diagnosis and prognosis of HCC in recent years, the underlying mechanisms of hepatocarcinogenesis and metastasis are still urgent to be elucidated. 2 MiRNAs bind to the 3′-UTR of their target mRNAs and subsequently result in repression of their translation or mRNA degradation, and numerous studies have revealed that abnormal expression of miRNAs plays a critical role in the process of carcinogenesis and tumor progression.3,8 In the present study, miRNA-1205 was identified as a tumor suppressor in HCC. In vitro and in vivo gain- and loss-of-function analyses demonstrated that decreased expression of miR-1205 significantly promoted HCC cell proliferation, whereas overexpression of miR-1205 resulted in opposite effects.
Among the multiple potential target genes of miR-1205 found by online prediction tools, CSNK2B was identified as a downstream effector of miR-1205. Negative correlation between their mRNA and protein levels was observed, and dual-luciferase reporter assays verified that miR-1205 directly binds to the 3ʹ-UTR region of CSNK2B mRNA, thereby inhibiting its’ transcription. As a regulatory subunit of CK2 kinase, several studies have reported that CSNK2B acts as an oncogene in various human malignancies such as gastric cancer, colorectal cancer, and HCC.20–22 In the present study, CSNK2B was revealed to be a main downstream effector of miR-1205 as evidenced by the cell biology experiments. Consistent with previous reports, CSNK2B significantly promoted HCC cell proliferation, and the effects of miR-1205 on colony formation ability were reversed by overexpression of CSKN2B, suggesting that the function of miR-1205 was mediated by suppressing CSNK2B transcription. This, to the best of our knowledge, was the first time CSNK2B was identified as a direct target gene of the miR-1205.
Although CK2 has been reported to promote aberrant activation of NF-κB pathway in HCC by phosphorylating IKK, IκBα, and p65,22,25 the mechanisms of CSNK2B itself in HCC remain largely unclear. Herein, we demonstrated for the first time that CSNK2B promotes CDK4 and p-Rb levels in HCC cells, suggesting that CDK4/p-Rb cell cycle pathway might mediate the oncogenic role of CSNK2B, and further colony formation assays verified this hypothesis by treatment with FDA-approved CDK4/6 inhibitor Palbociclib. Overall, the findings described above demonstrated that CSNK2B significantly enhanced CDK4 protein expression, indicating a functional link between oncogene CSNK2B and cell cycle regulation. However, this study has limitations, how exactly CSNK2B regulated CDK4 expression in HCC cells remains less clear. More in-depth research is needed to clarify the regulatory mechanisms and reveal whether CSNK2B exerts its function alone or through forming CK2 kinase.
Conclusions
The current work demonstrated that miR-1205 expression was reduced in HCC tissues, which possibly serves as an independent prognostic factor that predicts better survival of patients with HCC. Mechanistically, we found that miR-1205 inhibits HCC cell proliferation via the CSNK2B/CDK4 signaling axis. Our findings provide a novel theoretical basis and present a potential therapeutic target against HCC.
Supplemental Material
sj-docx-1-tct-10.1177_15330338221150544 - Supplemental material for MicroRNA-1205 Suppresses Hepatocellular Carcinoma Cell Proliferation via a CSNK2B/CDK4 Axis
Supplemental material, sj-docx-1-tct-10.1177_15330338221150544 for MicroRNA-1205 Suppresses Hepatocellular Carcinoma Cell Proliferation via a CSNK2B/CDK4 Axis by Xiang Li, MM, Shujie Xie, MD, Qin Xia, MM, Jia Yan, MM, Shuhuai Chen, MM, and Jia Shen, MM in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Data Availability
The data generated during the study are available from the corresponding author on reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Research Foundation of Hwa Mei Hospital, Ningbo Clinical Research Center for Digestive System Tumors, (grant number 2021HMKY16, 2019A21003).
Ethics Statement
Our study was approved by The Ethics Committee of Hwa Mei Hospital, University of Our study was approved by The Ethics Committee of Hwa Mei Hospital, University of Chinese Academy of Sciences (No. SL-NBEY-KY-2021-170-01, Date: 2021.11.11). The protocol was also supported by the Institutional Animal Care and Use Committee of Hwa Mei Hospital, University of Chinese Academy of Sciences and approved by the Guidelines for the Care and Use of Laboratory Animals provisions of administration and usage of laboratory animals by National Institutes of Health. 22 Each step was rigorously performed according to Declaration of Helsinki. All patients provided written informed consent prior to enrollment in the study.
Statement for the Animal Study Approval
Ethics approval of animal study was granted by the Institutional Ethics Committee of Hwa Mei Hospital, University of Chinese Academy of Sciences (No. SL-NBEY-KY-2021-170-01, Date: 2021.11.11). All animal experiments conformed with the ARRIVE 2.0 guidelines.
Statement for the Approval to Use Clinical Specimens
Ethics approval was granted by the Institutional Ethics Committee of Hwa Mei Hospital, University of Chinese Academy of Sciences (No. SL-NBEY-KY-2021-170-01, Date: 2021.11.11), and informed consent was signed by all patients enrolled in the present study.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.