
Abstract
Cholangiocarcinomas (CCAs) are a group of heterogeneous epithelial malignancies that can originate at the level of any location of the biliary tree. These tumors are relatively rare but associated with a high rate of mortality. CCAs are morphologically and molecularly heterogeneous and for their location can be distinguished as intracellular and extracellular, subdivided into perihilar and distal. Recent epidemiological, molecular, and cellular studies have supported that the consistent heterogeneity observed for CCAs may result from the convergence of various key elements mainly represented by risk factors, heterogeneity of the associated molecular abnormalities at genetic and epigenetic levels and by different potential cells of origin. These studies have consistently contributed to better defining the pathogenesis of CCAs and to identify in some instances new therapeutic targets. Although the therapeutic progress were still limited, these observations suggest that a better understanding of the molecular mechanisms underlying CCA in the future will help to develop more efficacious treatment strategies.
Keywords
Introduction
Cholangiocarcinomas (CCAs) are a heterogeneous group of malignant hepatic and extrahepatic tumors, located along the biliary tree. These tumors account for about 15% of all primary liver cancers and for about 3% of all gastrointestinal malignancies. According to their anatomical location, these tumors are classified as intrahepatic (iCCA, forming from proximal to second order bile ducts, ductules, and segmental ductules, located within the liver), perihilar (pCCA, arising outside the liver, at the level of large bile ducts located in the hepatic hilum and above the insertion of the cystic duct) and distal (dCCA, arising outside the liver, below the insertion of the cystic duct). iCCAs display 3 distinct growth patterns: mass-forming, periductal-infiltrating, and intraductal-growing; pCCA and dCCA display a different growth pattern, usually as nodular sclerosing tumors and less frequently as intraductal papillary tumors.
At the histological level, there are some remarkable differences between these tumors: pCCAs and dCCAs predominantly are represented by mucinous adenocarcinomas, while iCCAs display variable histology, with a small-type iCCA exhibiting a morphology resembling transformed interlobular bile ducts and a large-type iCCA resembling mucinous adenocarcinomas 1 (Figure 1).

Main subtypes of CCAs are subdivided into iCCA, pCCA, and dCCA and analyzed for their tumor location (lane 1, from the top to the bottom), histological features (lane 2) and putative cells of origin (lane 3).
CCAs are tumors displaying a considerable geographical variation, with an incidence remarkably higher in East Asian countries than in Western countries. A higher incidence of CCAs in Southeast Asia is due to infection with the fish-born liver fluke Opisthorchis viverrine, flat worm parasites: CCAs occurring secondary to fluke infestation may occur at the level of all the 3 anatomical sites of CCAs. 1 In the majority of countries, where liver flukes are not endemic, CCAs are less frequent malignancies. In Western countries, primary sclerosing cholangitis is a risk factor for CCA; among the other risk factors, cirrhosis and hepatitis B have a stronger association with iCCA, while bile duct stones are more related to pCCA and dCCA. A large meta-analysis carried out on the most relevant case-control studies published until 2020 identified risk factors for iCCA and eCCA. 2 The strongest risk factors for iCCA and eCCA are represented by biliary cysts and stones, cirrhosis, hepatitis B, and hepatitis C. 2 Particularly, choledoctal cysts were found to be most associated with both iCCA and eCCA; cirrhosis was more associated with iCCA than with eCCA; coledocholithiasis was more strongly associated with eCCA than with iCCA; in Eastern countries, cirrhosis and HBV was mor associated with a greater risk of iCCA than in Western countries. 2 In contrast to these findings, no significant associations were found between obesity and hypertension for either iCCA and eCCA. 2 Other studies showed that obesity, smoking and diabetes type II represent only weak/modest risk factors for the development of either iCCA or eCCA; at the level of inflammatory conditions, chronic pancreatitis represents a moderate risk factor for ICCA and a strong risk factor for eCCA; finally, nonalcoholic fatty liver disease (NAFLD) is a strong risk factor for iCCA and a moderate risk factor for eCCA 3 (Table 1).
Incidence, Risk Factors, Most Frequent Gene Alterations, and Cells of Origin of Various Types of Cholangiocarcinomas.

Abbreviations: iCCA, intracellular cholangiocarcinoma; eCCA, extracellular cholangiocarcinoma; cHCC-CCA, combined hepatocellular carcinoma and intrahepatic cholangiocarcinoma.
Molecular Abnormalities of CCAs
Recent studies have consistently contributed to define the molecular alterations observed in the different types of CCAs. These studies have shown the existence of significant differences at the level of molecular abnormalities of various CCA subtypes and have suggested also the possible different cellular origins of these tumors.
A fundamental study by Nakamura et al In 2015 showed the results of a whole-exome sequencing analysis on 260 Japanese patients with iCCA and eCCA. 4 Three most frequently mutated genes in these tumors were TP53, KRAS and ARID1A, followed by SMAD4, BAP1, PIK3CA, ARID2, and GNAS; the majority of genes most recurrently mutated were altered in < 5% of the cases. 4 Some genetic alterations, such as those involving FGFR1-3, IDH1, IDH2, BAP1, RNF43, and EPHA2 genes are much more frequently observed in iCCAs than in eCCAs, while ARID1B and ELF3 mutations and PRKACA and PRKACB fusions are more recurrent in eCCAs than in iCCAs. 2 Some genetic alterations, such as TP53, KRAS, ARID1A, and PIK3CA mutations and CDKN2A/B loss are shared between iCCA and eCCA. 4 Jusakul et al 5 explored the molecular abnormalities of 489 CCA cases, including patients from 10 different countries; the top driver genes mutated in these tumors were TP53, ARID1A, KRAS, SMAD4, and BAP1 and less frequently APC, PBRM1, ELF3, BRAF, IDH1, and BRCA2. Wardell et al 6 analyzed genomic features of 412 biliary tract cancer samples from Japanese and Italian patients; TP53, KRAS, SMAD4, and ARID1A were the genes most frequently mutated in these tumors; copy number analysis showed frequent chromosome 1q and 19q amplifications and chromosome 4p, 4q, 9p, and 14q deletions. The analysis of the profile of gene alterations showed some remarkable differences: epigenetic genes were more frequently mutated in ICC samples, whereas eCCAs contained more cell cycle mutated genes; ICC samples were significantly enriched in mutations involving IDH1 and BAP1 genes; eCCAs were significantly enriched for mutations in TP53, KRAS, SMAD4, and ERBB3. 6 Interestingly, this study showed that 8% to 12% of iCCAs are associated with pathogenic or possibly deleterious germline mutations: the most commonly mutated germline genes were BRCA1 and BRCA2 6 (Figure 2).

Main subtypes of CCAs are subdivided into iCCA, pCCA, and dCCA and analyzed for their histological features (lane 1, from the top to the bottom), etiological actors (lane 2), presence of preneoplastic lesions (lane 3) and more frequent gene mutations (lane 4).
Other studies have shown the molecular heterogeneity of eCCAs, showing some remarkable differences between pCCAs and dCCAs. The analysis of a large set of Chinese patients with eCCAs showed that: the frequency of PIK3CA, FAT4, KDM6A, MDM2, and TCF742 mutations was significantly higher in pCCA compared to dCCA, while the frequency of TP53 and KRAS mutations was clearly higher in dCCAs than in eCCAs. 7 Simbolo et al 8 explored 99 eCCAs and confirmed the existence of some remarkable differences in the genomic and transcriptomic profiles of pCCAs compared to dCCAs: particularly, pCCAs displayed more frequent KRAS mutations, whereas TP53 mutations were more frequent in dECCAs. The integrative molecular analysis of 186 eCCA samples further supported their consistent heterogeneity. 8 In fact, Montal et al 9 classified eCCAs into 4 subgroups according to transcriptome-based analysis: (i) the tumors classified in the Metabolic group (19%) exhibited a hepatocyte-like phenotype with activation of the transcription factor HNF4 and enrichment in gene signatures related to bile acid metabolism; (ii) the Proliferation group (23%) was characterized by frequent ERBB2 mutations/amplifications, mTOR signaling activation and enrichment of MYC gene targets; (iii) the Mesenchymal group (47%) was enriched by TGFβ signaling activation, epithelial-to-mesenchymal transition and poor survival; (iv) the Immune group (11%) was characterized by a more consistent lymphocyte infiltration, overexpression of PD1/PDL1, features compatible with a better response to immune checkpoint inhibitors.
As above discussed, iCCA exhibits differing etiologies worldwide, with the highest incidence in Southeast Asia. Therefore, it seemed of interest to explore the occurrence of some differences in the genomic landscape of iCCA in Eastern and Western patients. A higher burden of DNA repair mutations and frequency of patients with high tumor burden in the Asian iCCA patients compared with the Western patients was observed. 10 Asian patients displayed significantly higher frequency in KTM2D, BRCA1/2, KRAS, and DDR2 compared with Western patients; in contrast, Western patients had higher frequency of IDH1, IDH2, CDKN2A, and CDKN2B compared to Asian patients. 10
Integrative studies have contributed to define the heterogeneity of iCCAs. Different clustering methods have been used in these studies, leading to several molecular classifications of iCCAs. Sia et al 11 have identified 2 iCCA groups defined as proliferation and inflammation: the proliferation class was characterized by mutations in KRAS and BRAF, activation of oncogenic signaling pathways (RAS/MAPK, MET, EGF, and VEGF), DNA amplifications at the level of 11q13.2 and 14q22.1 chromosomal regions; the inflammation class characterized by activation of inflammatory signaling pathways, overexpression of cytokines and STAT3 activation. The proliferation class was associated with a negative prognosis.
Nepal et al 12 have reported the analysis of 496 iCCA patients by whole-exome sequencing and DNA methylation analysis and have stratified these patients into 4 different groups according to the presence of the 3 main mutated genes KRAS, TP53, and IDH1, thus defining a KRAS, TP53, IDH1, and an undetermined group. The KRAS group was characterized by SMAD4, ERBB, and VEGF alterations, by enrichment in immune-related pathways, and was associated with a negative prognosis. 12 The TP53 group showed a significant enrichment in PTEN, RAB1, MAPK, and WNT signaling. 12 TP53 mutation was found to be significantly associated with hepatitis B virus infection. The IDH group displayed the most extensive DNA methylation deregulation, frequent ARID1A, and BAP1 alterations, and metabolic pathway gene alterations. 12 The “undetermined” group was associated with extensive structural alterations, frequent ARID1A alterations, FGFR fusion, and mTOR pathway alterations; in 70% of these patients, the focal chromosomal amplification involving the gene methyltransferase-like 13 (METTL13). 12 A recent study showed a significant role of the transcription factor METTL3 in the promotion of iCCA progression; its targeting could be of therapeutical relevance. 13 The study of Nepal et al provided also a detailed analysis of structural variations and copy number alterations occurring in iCCAs: the most frequent structural variation is represented by FGFR gene fusion observed in 6% to 14% of iCCAs; recurrent driver gene amplifications involved ERBB2 (2%-12%). MDM2 (0%-13%), EGFR (1%-16%), and CCND1 (10%-13%); deletion of the gene locus including genes CDKN2A and CDKN2B is observed in 10% to 20% of iCCAs. 13
iCCAs can be distinguished into 2 different histopathological subtypes following the size of the affected bile ducts: small duct and large duct types. The small duct type accounts for 36% to 84% of iCCAs and was defined as a peripheral ductular and cholangiolar type and is characterized by the presence of tubules of small-sized cuboidal or low-columnar tumor epithelial cells with no mucin production; these tumors are frequently associated with B or C viral hepatitis and nonalcoholic steatohepatitis. 14 The large duct type accounts for 6% to 60% of iCCA cases, is defined as a large duct type, and is characterized by the proliferation of tubular or gland-like structures composed of mucin-producing columnar cells and usually shows a highly aggressive growth pattern accompanied by a desmoplastic reaction; these tumors are frequently associated with parasitic infection in bile ducts, hepatolithiasis, and primary sclerosing cholangitis. 15 The remarkable differences existing between small and large bile duct iCCAs are supported also by immunohistochemical and molecular studies: large duct iCCAs are characterized by diffuse immunoreactivity to S100P and displayed higher serum levels of CA 19-9 and CEA than small duct iCCAs; small duct iCCAs exhibited reactivity to N-cadherin and/or NCAM. 16 Intracellular and extracellular mucin is clearly abundant in the large duct type, while is absent or only scarcely expressed in the small duct type. C-reactive protein (CRP) is a marker allowing to differentiate of small duct from large duct types since its positivity is limited to small duct type. 17 Inflammation and proliferation-related markers differentiate small duct type from large duct type, the former one being preferentially expressed in small duct type and the latter one in large duct type. 18
KRAS mutations were more frequent in large bile duct iCCAs, while IDH mutations and FGFR2 translocations were restricted to small bile duct iCCAs. 15 Loss of BAP1 expression and IDH1 mutations were restricted to the small bile duct iCCA subtype, while about one-third of large bile duct iCCA lacked the expression of SMAD4. 19 Large duct type iCCAs display frequent mutations in TP53, KRAS, and some genes of the TGFβpathways, such as SMAD4, TGFBR2, FBXW7, and MYC. 19
These findings support different pathogenetic mechanisms for small bile duct and large bile duct iCCA: chronic hepatitis or cirrhosis induce the formation of small duct iCCA promoting the neoplastic transformation of a cell component present in small bile ducts, such as hepatic stem or progenitor cells or cuboidal cholangiocytes; chronic biliary inflammation caused by liver fluke infection or primary sclerosing cholangitis induce neoplastic transformation of some cellular elements present in the large bile ducts. 20
Interestingly, recent single-cell transcriptomic studies have supported the existence of 2 molecularly distinct subtypes of iCCA corresponding to small and large bile duct types: SP100P is a marker for large bile duct type, while SPP1 is a marker for small bile duct type. 21 SPP1+ iCCAs have reduced levels of infiltrating CD4+ T cells, CD56+ NK lymphocytes and increased CCL18+ macrophages and PDL1+ CD8+ T cells; in these tumor cells, SPP1 expression is promoted by the transcription factor CREB3L1. 20 SPP1+ iCCAs display significantly more SPP1+ macrophage infiltration and are composed by tumor cells at different stages of differentiation, such as elements ALB+ (hepatocyte differentiated) and ID3+ (with stemness properties). 21
Very recent integrated analyses of molecular abnormalities and of transcriptomic or proteomic profiles strongly support a consistent heterogeneity of iCCA, whose definition is fundamental for a better understanding of the origin, development, and progression of these tumors and for the identification of new potential therapeutic targets. Dong et al performed a proteogenomic characterization of 262 iCCA Chinese patients: 16 frequently altered driver genes were identified, including TP53, KRAS, FGFR2, IDH1, IDH2, CAP1, ARID1A, and PBMR1; 3.5% of the tumors displayed high tumor mutational burden; KRAS mutations were mutually exclusive IDH1, IDH2, BAP1, and FGFR2 alterations, while FGFR2 alterations were mutually exclusive with TP53, KRAS, IDH1, and IDH2 mutations. 22 Proteogenomic analysis showed that iCCAs with TP53, KRAS, BAP1, IDH1, or IDH2 driver mutations were associated with distinct profiles of activation or inhibition of specific biochemical pathways: TP53 mutations were associated with up-regulation of cell-cycle, drug metabolism, phagosome, and carbon metabolism pathways; KRAS mutations were associated with increased expression of proteins involved in the cell-cycle pathway; BAP1 mutations were associated with increased expression of proteins involved in ECM, bile secretion, phagosome, and inflammation; IDH1 and IDH2 mutations were associated with increased expression of proteins related to ECM, bile secretion and MAPK activation. 22 FGFR2 fusions represent a distinct subgroup of iCCAs and display activation of the Rho GTPase pathway and the generation of neoantigen candidates for personalized immunotherapy. 22 According to the protein pattern of expression, through clustering, 4 subgroups were identified: S1, characterized by abundant expression of inflammatory proteins, KRAS mutations, and intrahepatic metastasis; S2, characterized by high expression of proteins related to cancer-associated fibroblasts and ECM, frequent KRAS, TP53, and FGFR2 alterations, frequent lymph node metastases; S3, characterized by elevated MAPK and metabolic proteins, TP53 mutations and frequent HBV infection; S4, characterized by maximum expression of adhesion and biliary-specific proteins, BAP1, IDH1, and IDH2 mutations and FGFR2 alterations and lower level of metastases. 22
Wang et al 23 have performed a genomic analysis of 1481 iCCAs derived from different populations and have defined the profile of co-occurrence or mutual exclusivities among recurrent driver mutations. The 7 most frequent driver mutations (TP53, KRAS, SMAD4, IDH1, IDH2, BAP1, and FGFR2-fus) displayed a pattern of pair-wise co-occurrences or mutual exclusivities, compatible with their aggregation into 3 different clusters: Cluster 1, subdivided into cluster 1A with KRAS mutations and cluster 1B with TP53/SMAD4 mutations, displayed a large bile cell morphology, high CA 19-9 levels and a negative prognosis; Cluster 2, subdivided into 3 subgroups, characterized by either IDH1, IDH2, or BAP1 mutations or FGFR2-fus, displayed a small bile duct morphology, low CA 19-9 levels and better prognosis; Cluster 3, characterized by the absence of one of the driver mutations, displayed in 43% of cases mutations of chromatin modifiers or/and of RTK/PI3 K, cell-cycle, DNA damage repair genes and in the remaining 57% of cases none of these mutations. 23
The analysis of the microenvironment, together with genomic data, allowed classification of iCCAs into 5 classes. 19 These 5 classes pertain to 2 different profiles: an inflamed (35%) and a noninflamed (65%) profile. The inflamed classes were named immune classical (10%) and inflammatory stroma (25%): particularly, the inflammatory stroma subtype exhibits T cell exhaustion, abundant stroma, and KRAS mutations. 19 Within the noninflamed classes: the desert-like class (20%) is characterized by the lowest immune infiltration, with the presence of an abundant cellular component represented by regulatory T cells; the hepatic stem-like class (35%) is characterized by frequent mutations of BAP1 and IDH1/IDH2 and FGFR2 fusions and by the presence of M2-like macrophages; the tumor classical (10%) is defined by cell-cycle pathways and is associated with a poor prognosis. 24
Another recent study evaluated the tumor microenvironment of iCCAs and defined 4 immune subtypes. 25 The I1 subtype displayed an immune desert pattern characterized by weak expression of all signatures associated with the tumor microenvironment, with strong attenuation of tumor and stromal immune signaling; the I2 subtype is characterized by high expression of inflammatory and immune checkpoint pathways and of activated fibroblasts, with overexpression of major histocompatibility complex class I and class II molecules, immune checkpoint molecules, regulators of macrophage activity; the I3 subtype is characterized by the strong expression of M2-polarized macrophage signature and by low expression of lymphoid signatures; the I4 subtype displayed mesenchymal features with high expression of fibroblast-associated signatures. 25
A recent study reported the integrative genetic analysis of 454 samples of biliary tract cancers and showed the subdivision of these tumors into 4 different clusters according to their mutational profile and, in parallel through gene expression studies showed that these 4 clusters differed for their immune signatures: cluster 1 corresponded to tumors of all 3 biliary cancer subtypes (gallbladder, iCCA, and eCCA), displayed 3 commonly altered genes (TP53, KRAS, and ATM) and the highest PD-L1 expression and exhibited the most activated immune signature, with most inflamed tumor immune microenvironment and increased B cells and plasma cells; cluster 2 corresponded to all 3 biliary cancer subtypes, displayed frequent CDKN2A, CDKN2B, TP53, and KRAS gene alterations and exhibited an immune signature characterized by decreased expression of memory resting CD4 cells; cluster 3 corresponded to all 3 biliary cancer subtypes, showed frequent ARID1A, PBRM1, and IDH1 gene alterations and an immune signature characterized by increased expression of activated NK cells and M2 macrophages and decreased B cells; cluster 4 was enriched for iCCA, displayed frequent BAP1 and FGFR2 gene alterations, low PD-L1 expression and an immune signature characterized by decreased activated dendritic cells. 26
Combined HCC and ICC (cHCC-ICC) is a rare type of liver cancer exhibiting both hepatocellular and biliary epithelial cell differentiation. According to anatomopathological criteria, cHCC-ICCs are subdivided into 3 subtypes: (i) separate subtype where different lesions are physically distinct and exhibit different histology; (ii) combined subtype where defined and separated areas of HCC and ICC components are located in the same tumor; (iii) mixed subtype, where HCC and ICC components are intimately mixed within the same tumor.
The molecular characterization of this tumor was made more complex by its rarity and by the presence of 2 different histological components. In a pivotal study, Moeini et al 27 reported in 2017 the results of an integrative analysis of 18 cases of cHCC-ICC, based on gene expression profiling, DNA copy number analysis, and exome sequencing. This analysis showed that a part of cHCC-ICC corresponds to a peculiar distinct biliary-derived tumor, defined as cholangiolocellular carcinoma (CCLC). This tumor was NCAM-positive, chromosomally stable, and displayed TGFβ signaling pathway upregulation and enrichment of inflammation-related and immune response signatures. 27 The rest of cHCC-ICCs corresponded to the classical subtype with common lineage for HCC and ICC components; TP53 was the gene most frequently mutated in these tumors. A significant correlation was observed between the CNAs of the HCC and of the iCCA components of the tumors, suggesting a monoclonal origin. 27
CCLC is a variant of small duct type iCCA composed of cuboidal cells forming cords or ductal-like structures and is associated with a better prognosis compared to conventional small duct type. 28 The term cholangiolo is related to the cholangioles or canals of Hering. 28 Clinicopathological studies have suggested that CCLC may represent a subtype of combined hepatocellular–cholangiocarcinoma, 29 while the above-reported studies support that CCLC is a part of iCCA. 28 It is important to note that CCLC can be distinguished from conventional small duct type iCCAs for its better outcomes, as evidenced by longer OS and PFS. 30
A study by Joseph et al 31 reported the analysis of 20 cHCC-ICC cases by deep targeted sequencing and showed that these tumors display frequent TP53 and TERT promoter mutations; IDH1, IDH2, BAP1 mutations, and FGFR2 fusions were more rarely observed in these tumors compared to iCCAs.
Wang et al 32 reported the analysis of the mutational profile of the HCC and iCCA components of 7 cHCC-ICC samples, providing evidence that the 2 components display from 33% to 86% of private mutations, suggesting a high degree of tumor heterogeneity; however, numerous mutations are shared by HCC and iCCA components, suggesting the monoclonal origin of these tumors. This study showed also a high expression of EpCAM in these tumors, a finding suggesting the stemness of cHCC-CCAs. 32
In 2019, Xue et al 33 reported the results of a fundamental study representing the largest exploration of cHCC-ICC patients, involving the analysis of 133 cases (6 separate, 56 combined, and 59 mixed). This study involved whole-exome sequencing, whole-genome sequencing, and RNA sequencing. TP53, TERT promoter, AXIN1, KMT2D resulted to be the most frequently genes. The genomic comparison of cHCC-ICC with HCC and iCCA showed some remarkable differences: in cHCC-ICC TP53 mutations are more frequent than in HCC and iCCA; TERT promoter mutations are less frequent in cHCC-ICC than in HCC, while these mutations are absent in iCCA; KRAS, RAP1, IDH1, and IDH2 mutations are markedly less frequent in cHCC-ICC than in iCCA; AXIN1 mutations are observed in mixed but not in combined cHCC-ICC. 33 Primary liver cancers were subdivided into 4 transcriptional subtypes (P1 to P4): cHCC-ICCs were enriched in P1 and P2. Epithelial-mesenchymal transition (EMT) is the most enriched gene set in P1, whereas xenobiotic metabolism and bile acid metabolism are the most enriched gene sets in P2; cluster P1 was particularly observed in combined cHCC-ICC, while cluster P2 was frequently in mixed cHCC-ICC. 33 These observations supported the important conclusion that combined and mixed types of cHCC-ICC are distinct molecular subtypes, with the combined subtype displaying more similarity to iCCA and the mixed subtype being more similar to HCC. 32 Bulk and single nucleus sequencing studies supported the conclusion that while separate cHCC-ICCs are either of monoclonal or polyclonal origin, mixed and combined cHCC-ICCs are always of monoclonal origin. 27 In spite of their molecular differences, mixed and combines cHCC-ICC subtypes displayed stem-like features: this property seems to be related to the reduced repression exerted by mutant TP53 on the cellular plasticity, as supported by the elevated expression observed in these tumors of Nestin, a marker of bipotential liver progenitor cells. 33
Murugesan et al 34 have explored the genomic profiling of 73 cHCC-ICC cases; they used a machine learning genomics-driven model to classify cHCC-ICCs into HCC-like and iCCA-like: 16.4% of these tumors were classified as iCCA-like, 57.5% as HCC-like and 26.3% as ambiguous. The HCC-like cases displayed genomic alterations in TERT (71.4%), CTNNB1 (9.5%), and MYC (9.5%), whereas iCCA-like cases were TERT, CTNNB1, and MYC wild-type and exhibited genomic alterations in ARID1A, FGFR2, and IDH1 (all 25%). 34
Some recent studies have explored the association between molecular abnormalities of BTCs with response to chemotherapy. Yoon et al 18 showed favorable responses to chemotherapy. Were preferentially observed in the small duct type compared to the large-duct type of iCCA; tumors exhibiting TGF-β family and DNA damage response pathway alterations displayed poor response to chemotherapy. Boerner et al 35 have investigated 412 iCCA cases at the level of genomic alterations by NGS analysis and have explored the possible associations between these alterations and clinicopathologic variables and outcomes. For all patients, TP53, KRAS, and CDKN2A alterations predicted poor overall survival; these alterations were enriched in cases with advanced disease. In surgically resected patients, TP53 mutations and CDKN2A deletions independently predicted shorter OS and high-risk clinical variables. 35 Furthermore, TP53 mutations, KRAS mutations, and CDKN2A deletions similarly predicted worse outcomes in patients with unresectable tumors. 35
Epigenetic Mechanisms in CCA Development
Epigenetic mechanisms play an important role in the initialization and development of CCA and contribute in a relevant way to the tumor phenotype. As above discussed, deletions and mutations of genes encoding the chromatin remodeling genes BAP1, ARID1A, and PBRM1 and gain-of-function mutations of IDH1 and IDH2 genes are the most common alterations perturbing the epigenetic landscape of iCCA. The deregulation of several epigenetic pathways, mainly represented by DNA methylation, histone modifications, and abnormal expression of noncoding RNAs induce aberrant transcription and gene expression, thus contributing to the development of CCA and of its malignant phenotype. 36 Several studies have shown a deregulated regulation of DNA methylation in CCA leading to the formation of deregulated DNA methylation motifs in CCA cells compared to their normal counterpart: one of these prevalent abnormalities is represented by hypermethylation of multiple. DNA CpG sites, occurring at the level of promoter-associated regions nonrandomly distributed and enriched for genes involved in cancer-related pathways, including WNT, TGFβ, and PI3 K signaling pathways. 37
An integrative genetic and epigenetic analysis carried out in 52 iCCA patients with a nonliver fluke-associated etiology identified 4 groups with prognostic relevance: (i) An IDH group characterized by IDH1 and IDH2 mutations, a unique pattern of high methylation values, high levels of CNAs and high values of latent methylation component C2 (LMC2); the high level of methylation observed in this group is related with the neo-enzymatic activity of mutated IDH, with consequent inhibition of TET2 activity and inhibition of active DNA demethylation. (ii) The H group if characterized by high methylation levels, frequent CNAs, and high LMC4 levels. (iii) The M group is characterized by a mixed LMC composition, low frequency of deletions, and specific gain of the chromosome arm 8q harboring MYC gene. (iv) The L group is characterized by the highest proportion of LMC1 and LMC5, low mutational frequency, and low methylation levels. 38
The analysis of the DNA methylation profile in CCA allowed also us to identify of some new biomarkers with a high diagnostic capacity. In this context, a recent study allowed us to identify HOXD8 hypermethylation as a highly sensitive and specific biomarker for BTCs in both tumor tissues and in bile samples. 39
Some studies have shown the occurrence of alterations of histone deacetylases (HDACs), enzymes involved in chromatin remodeling through the deacetylation of lysine residues. Thus, a recent study showed that HDAC1 is significantly overexpressed in CCA and the suppression of its activity using the drug inhibitor JSL-1 reduces progression and metastasis in CCA experimental models, through the TPX2/Snail axis. 40
Micro-RNAs are small noncoding RNAs that play an important role in the control of gene expression acting through posttranscriptional mechanisms. The expression of many micro-RNAs is deregulated in CCA: some of these deregulated micro-RNAs act as oncogenes or tumor suppressors in CCA development and their targeting may offer new therapeutic opportunities. 41
Cellular Origin of Biliary Cancers
The cellular origin of biliary cancers remains highly debated. However, in spite of this consistent incertitude, the available evidence supports a different cellular origin of iCCAs compared to eCCAs.
Various criteria can be adopted for the assessment of the cellular origin of cancer. Particularly studies on the characterization of genetic alterations, histological features, and animal models represent important tools to unveil the cellular origin of cancer cells.
The advances in genomic studies have provided a large set of data driving a better understanding of the molecular mechanism driving cholangiocarcinogenesis. The progresses in molecular studies have provided strong support for the development of CCA preclinical models suitable for: (i) investigating the molecular mechanisms involved in cholangiocarcinogenesis from tumor initiation to tumor progression and metastatization; (ii) identifying druggable molecular targets and diagnostic/prognostic biomarkers; (iii) evaluating the effect of new drugs or drug combinations, in an attempt to develop more efficient therapies; (iv) to decipher the cells of origin of various types of biliary tract cancers. 42
The existence of marked geographical and etiology heterogeneity of iCCA and eCCA suggested the occurrence of different cell-initiating tumors in these tumors.43,44 Particularly, concerning iCCA 2 cells of origin have been hypothesized (mucin-secreting cholangiocytes or hepatic progenitor cells) to explain the consistent histological and molecular heterogeneity of these tumors 34 ; pCCA and dCCA were proposed to originate from mucin-secreting cholangiocytes.43,45
The identification of the cell type of origin of cancer is of fundamental importance. The cell of origin of cancer can be determined through a comparative analysis of the distribution of mutations and chromatin organization of specific cell types since mutations are more likely to occur in transcriptionally active chromatin. 46 A prediction of the cell-of-origin of biliary tract cancers was performed, as based on the analysis of somatic mutation profiles and epigenetic features, showing that 33% of biliary cancers were classified as hepatic and 38% as epithelial cell types: iCCA samples were more frequently classified as hepatic (43.5% and 17.4%), while eCCA samples were more frequently classified as epithelial (0% and 83.3%). 6
A recent study of characterization of the genomic landscape, englobing a large set of iCCA and pCCA patients supported the existence of several remarkable genetic differences between these 2 groups of patients. 37 Thus, the nonsynonymous tumor mutation burden was significantly higher in iCCA (0.61/Mb) than in pCCA (0.47/Mb); furthermore, iCCA had a higher copy number alteration burden than pCCA (25% vs 8.4%). 40 pCCAs with a high copy number tumor burden are associated with a poor prognosis. Several genes displayed different mutation rates between iCCA and pCCA: TP53, ARID1A, PBRM1, MACF1, EPHA2, ARID2, IDH1, PTEN, RB1, BRAF, NRAS SLC8A1, AXIN, MLLT4 were iCCA-enriched genes, while RBM10, TGFBR2, PIK3R1, ELF3, NACC1, METTL14 were pCCA-enriched genes. 47 MTTL14 encloses a methyltransferase and exerts a tumor-suppressive effect.
Histological studies support the origin of pCCAs and dCCAs from columnar mucous cholangiocytes and perihilary glands (PBG, located around the extrahepatic and perihilar bile ducts). There is evidence that eCCAs may derive from preneoplastic lesions originating from surface epithelium or from PBGs. 48 In 2012, Nakanuma and Sato provided evidence PBGs contain cells involved in the origin of intraductal papillary neoplasms of the bile duct (IPNB) and early neoplastic lesion of invasive cholangiocarcinoma. 49 Immunohistochemistry and gene expression studies have shown a consistent similarity to the mucin-producing cholangiocytes lining hilary bile ducts and to PBGs. Other studies have confirmed a possible origin of cystic and papillary neoplasms from the perihilary glands. 50
Studies carried out in primary sclerosing cholangitis have shown an involvement of PBG in biliary duct inflammation with fibrosis: peribiliary gland hyperplasia associated with tissue fibrosis and dysplastic lesions; the presence of cells with enhanced expression of Hedgehog component in PBG suggests the presence in these glands of activated biliary stem cell populations. 51 Other studies have shown that CCAs emerging in primary sclerosing cholangitis patients are mucin-producing tumors characterized by PBG involvement and a high expression of stem/progenitor cell markers. 52 It was estimated the cumulative risk for CCA development in primary sclerosing cholangitis of 6% to 22%. A recent meta-analysis based on a large set of data showed that individuals with primary sclerosing cholangitis have a markedly increased risk of developing hepatobiliary tumors compared to that of individuals without primary sclerosing cholangitis, evaluable as a relative risk of 584 for CCA, 155 for hepatobiliary cancer and 30 for liver cancer. 53 Goeppert et al 54 reported the genomic characterization of 186 primary biliary cancers occurring in patients with primary sclerosing cholangitis; regardless, or the anatomical location, these tumors exhibited a uniform molecular and histological characteristics resembling eCCAs. Particularly, a high frequency of TP53 (35.5%), KRAS (28%), CDKN2A (14.5%), SMAD4 (11.3%), PIK3CA (9.1%), CDKN2B (8.6%), ERBB2 (8.1%), KM5A/KM6A (7%), ROBO1 (7%) was observed; FBW7, TGFBR2; GNAS, SMARCA4, BRAF, and ARID1A genes were less frequently mutated (2%-5%); CNAs were observed primarily inn CDKN2B (7.5%), CDKN2A (7%), and ERBB2 (4.3%); FGFR translocations were absent, as well as BRACA1/2 mutation and IDH1 mutation was very rarely observed in these tumors. 54 furthermore, a high rate (18.3%) of precursor lesions was observed in these tumors. 54
Recent studies have characterized IPNBs at the cellular and molecular levels. These premalignant neoplasms show the intraductal growth of biliary epithelium with papillary or villous features. IPBNs are associated in 50% of cases with an invasive CCA. IPNBs have been classified as type 1 or type 2: type 1 IPNBs are characterized by a more homogeneous morphology than type 2, and their architecture is represented by regular structures with papillary, villous, or tubular morphology and usually display a low degree of cellular dysplasia and high mucin accumulation; type 2 IPNBs are characterized by a more irregular architecture with irregular papillary, villous or tubular structures and usually display a high degree of dysplasia.55,56 IPNBs display either intrahepatic or extrahepatic localization. Two recent studies have explored the mutational profile of IPNBVs, showing some remarkable differences between type 1 and type 2 IPNBs: type 1 IPNBs have a higher mutation frequency of KRAS, GNAS, and RNF43 genes; type 2 IPNBs exhibit a higher rate of TP53 and SMAD4 mutations.57,58 A part of IPNBs do not show the predominant typical papillary morphology, but display tubular growth of epithelial elements with low mucin production: these cases have been defined as intraductal tubulopapillary neoplasms (ITPNs). 59 ITPNs are mostly localized intrahepatically. A recent study showed remarkable differences in the mutational profile of IPNBs compared to ITPNs: IPNBs exhibited mutational profiles of eCCAs, with predominant TP53, SMAD4, and KRAS mutations; ITPNs displayed a lower number of mutations. 60 According to these findings, it was proposed that IPNBs and ITPNs may represent a peculiar intermediate form of intrahepatic and extrahepatic cholangiocarcinogenesis. 60 Interestingly, some IPNBs and ITPNs may represent a precursor lesion for iCCA of the small-duct type.
Some animal models of eCCAs further supported different genetic driving mechanisms compared to those observed in iCCAs. The cholangiocytes lining the extrahepatic biliary tree have a peculiar embryological origin in that they originate together with the ventral pancreas and do not derive from the liver; these cells originate from the differentiation of PDX1-positive progenitor cells located at the level of the caudal part of the ventral endoderm. 61 Interestingly, this PDX1-positive extra-hepatic epithelium is very susceptible to the oncogenic transformation by activated mutant PIK3CA, but resistant to the oncogenic effects induced by mutant KRAS. 62 Using suitable models of eCCA oncogenesis in mice it was shown that the level of PIK3CA activation and repression of p27 are key oncogenic determinants; p27 loss promoted extrahepatic bile duct oncogenesis in cooperation with KRAS mutations. 62
As above discussed, histological studies have strongly supported the hypothesis that cholangiocytes present within the intrahepatic small bile ducts are considered the cells of origin of small duct type iCCA, while column cholangiocytes or perihilary glands are considered the cells of origin of large duct iCCA.
The use of genetically engineered mouse models (GEMMs) and lineage tracing systems represent a fundamental tool for the evaluation of the cell of origin of cancer. The use of some of these mouse models and of lineage tracing systems have supported, at variance with histological evidence, a cellular origin of iCCA from the malignant transformation of hepatocytes. 63 Several of these studies have shown that the specific and selective activation of NOTCH in hepatocytic cells alone or in cooperation with AKT signaling activation64,65 or with TP53 mutations 66 that induce loss-of-function induce the malignant transformation of hepatocytes into neoplastic biliary cells. In this context, particularly relevant was the study carried out by Sekiya and Suzuki 67 using a model of experimental iCCA by chronic liver injury: in this model, hepatocytes were labeled with heritable, cell type-specific reporters; in these mice, iCCAs are generated by biliary lineage cells derived from hepatocytes; NOTCH signaling activation was critical for hepatocyte conversion into biliary lineage cells.
Other animal models based on GEMMs have supported a cell origin of iCCAs from bile duct cells. In an initial study, O’Dell et al 68 reported the development of a GEMM model based on combined TP53 inactivation and KRAS activation leading to the generation of iCCA, mimicking at histologic and molecular levels the human disease. Hill et al 69 showed that iCCA driven by KRAS and TP53 may originate from both mature cholangiocytes and hepatocytes and factors such as chronic liver injury determine the cellular path of progression and the resulting cancer phenotype: in fact, the introduction of KRAS and TP53 mutations into cholangiocytes resulted in iCCA generation, emerging from premalignant biliary intraepithelial neoplasia lesions, while mature hepatocytes are resistant; however, liver injury accelerated hepatocyte-driven tumorigenesis and promoted a phenotypic switch to iCCA.
The relevant cooperation of TP53 and KRAS in promoting the development and the malignancy of iCCA is directly supported by a recent study reporting a molecular and clinicopathologic analysis on 3 large cohorts of iCCA patients extensively characterized at molecular and clinical levels: TP53 and KRAS mutations were associated with a poor prognosis and this phenomenon was particularly evident for double mutant (TP53/KRAS) patients. 70
Guest et al 71 reported the development of an iCCA model based on chronic liver inflammation and transfection with mutant TP53 leads to the development of iCCA generated from cholangiocytes: in these tumors, the induction of NOTCH3 is required for oncogenic transformation of cholangiocytes. Interestingly, NOTCH3 was found to be the NOTCH family receptor mostly overexpressed in human iCCAs.
Other animal models based on the study of the in vivo effects of PTEN loss and KRAS activation on cholangiocarcinoma development in mice supported a biliary tract origin. In this context, Marsh et al 72 reported the results of a study based on an inducible Cre-LoxP-based approach to coordinately delete PTEN and activate KRAS within the adult mouse biliary epithelium. Activation of KRAS alone had only limited effects on biliary epithelium, while PTEN loss elicited the development of low-grade neoplastic lesions, following a long latency; however, the combination of both mutations induces the rapid development of proliferative biliary lesions, progressing from dysplasia to invasive carcinoma. 65 These observations supported the conclusion that PTEN loss and KRAS activation create a permissive environment in biliary epithelial cells for cholangiocarcinoma development. 72 A second study explored the effect of the introduction in embryonic hepatic bipotential progenitor cells (hepatoblasts): liver-specific KRAS activation in combination with homozygous PTEN deletion cooperate to induce iCCAs exclusively; KRAS activation and heterozygous PTEN deletion in combination induced the formation of both iCCAs and HCCs; KRAS activation alone induced the formation of HCCs alone. 73 A cell lineage tracing system showed that iCCAs originate from cholangiocytes but not from hepatocytes. 73
The development of another model of cholangiocarcinogenesis driven by PTEN loss helped to define the possible origin of cHCC-ICCAs. Thus, Chen et al 74 reported the development of a liver mouse model based on the deletion of PTEN in SOX9+ liver progenitor cells: the tumor cells generated by this approach generate mixed hepato-CCAs; liver injury causing a steatotic condition induces an increased tumor incidence and accelerate tumor formation. These observations suggest that transformed SOX9+ are able to act as tumor-initiating cells and that cHCC-CCAs may derive from the malignant transformation of bipotent SOX9+ progenitor cells. 74
Recent Developments in Targeted Therapy of CCAs
CCAs represent a group of highly heterogeneous tumors that differ in their anatomical location, epidemiological distribution, risk factors, molecular abnormalities, and potential cells of origin. The studies carried out in the last years have consistently contributed to defining this heterogeneity and have provided a characterization of the genomic alterations observed in these tumors.
These studies were extremely relevant not only for their contribution to understanding the molecular pathogenesis of CCAs, but also because have led in a subset of these tumors to the identification of molecular abnormalities that can be selectively targeted at the pharmacological level (Table 2). Particularly, 2 genetic alterations detected in iCCAs are amenable to targeted therapy: FGFR2 fusions and IDH mutations. As above shown, 10% to 15% of iCCAs display FGFR2 genomic alterations, mostly related to gene fusions (10%-12%) and more rarely to gene mutations and amplifications. Clinical studies with agents targeting FGFR2 have shown consistent clinical responses. Pemigatinib, a tyrosine multikinase inhibitor that blocks FGFR1-3, showed significant clinical activity in phase I and II clinical studies involving previously treated iCCA patients with FGFR-fusions or rearrangements 75 ; importantly, second-line treatment with pemigatinib may be associated with longer progression-free survival compared with second-line treatment with systemic therapy received before study enrollment: this finding, however, needs to be confirmed in a prospective study. 76 Patients with co-occurring gene alterations at the level of BAP1, CDKN2A/B, TP53, PBRM1, ARID1, or PTEN had shorter median progression-free survival. 77 In 2020 the US Food and Drug Administration (FDA) and in 2021 the European Commission approved pemigatinib for the treatment of patients with previously treated advanced iCCA patients with FGFR2 fusions or rearrangements. An ongoing phase III international trial is evaluating the treatment with pemigatinib versus platinum-based chemotherapy as first-line therapy for unresectable iCCA with FGFR2 fusions or rearrangements. 78
Clinical Trials Involving the Treatment of Patients With Advanced CCA With FGFR or IDH Inhibitors.
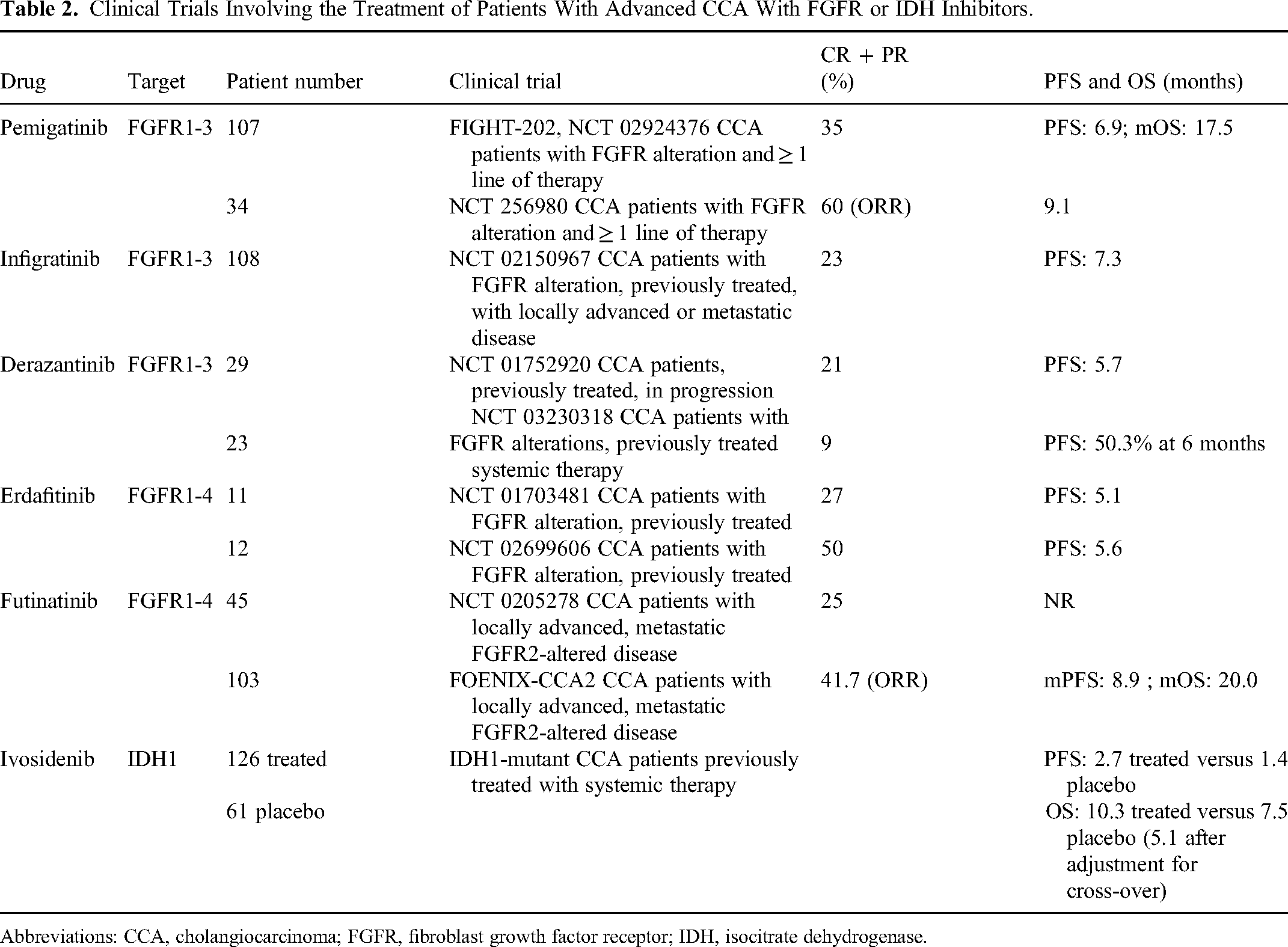
Abbreviations: CCA, cholangiocarcinoma; FGFR, fibroblast growth factor receptor; IDH, isocitrate dehydrogenase.
Pemigatinib represents the first molecularly targeted therapy to be approved for the treatment of CCA. However, several issues concerning the use of pemigatinib in the treatment of CCA still remain unresolved, such as the emergence of polyclonal mutations determining resistance to this drug and the identification of biomarkers able to predict the response to this FGFR inhibitor. 79
Infrigatinib is another FGFR1-3 inhibitor that was approved in 2021 by the FDA for the treatment of unresectable locally advanced, or metastatic CCA with FGFR2 or other rearrangements on the basis of the results of phase II clinical study carried out on 108 CCA patients with FGFR2 fusions or other rearrangements who had previously treated with a gemcitabine-containing regimen, and after a median follow-up of 10.6 months, there was an overall response rate of 23%, with 1 complete response and 24 partial responses. 80 A higher response rate was observed among patients treated in the second-line with infigratinib compared to those treated in the third or later lines of treatment. 80 PROOF 301 (NCT. 03773302), a phase III, randomized study of infigratinib versus gemcitabine plus cisplatin in patients with advanced CCA with FGFR fusion/rearrangement is ongoing.
Another FGFR1-4 inhibitor, futibatinib, received a breakthrough therapy designation by FDA for the treatment of CCA patients with FGFR2 rearrangements, based on the results of the FOENIX-CCA2 trial, showing an overall response rate of 34% and a disease control rate of 76%. 81 An updated analysis of the results of the FOENIX-CCA2 trial confirmed in 103 CCA patients with FGFR2 fusions/rearrangements, previously treated, an ORR of 41.7% and a duration of response of 9.7 months, with a PFS of 8.9 months and a mOS of 20.0 months. 82 Other observations further supported the efficacy of futibatinib in the treatment of CCA patients with FGFR2 fusions/rearrangements. Thus, indirect treatment comparison of futibatinib with chemotherapy and pemigatinib in CCA patients with FGFR2 fusions/rearrangements suggests that futibitib provides longer survival versus chemotherapy; similar efficacy was observed for futibatinib compared with pemigatinib. 83 Meric-Bernstam et al 84 have reported the results of a phase I dose-expansion trial evaluating the response of advanced tumors with FGFR1-3 aberrations to futibatinib. The greatest anti-tumor activity was observed in FGFR2 fusion/rearrangement-positive iCCA, with 25.4% of the overall response rate. 77 Some responses were observed also among patients previously treated with other FGFR inhibitors. 85
About 15% to 20% of iCCA patients displayed mutant IDH1/IDH2 genes; iCCA cells with IDH1/IDH2 mutations markedly accumulate the metabolite 2-hydroxyglutarate (2-HG): increased 2-HG levels induce inhibition of methylcytosine hydroxylases and histone demethylases, with consequent deregulation of transcription of some genes. Ivosidenib is an oral, small-molecule inhibitor of mutant IDH1 and it was approved in 2021 by FDA for the treatment of iCCA patients previously treated, locally advanced, or metastatic with an IDH1 mutation. The approval was based on the results of phase III clinical trial ClarlIDHy trial enrolling 187 patients with IDH1-mutant iCCAs, previously treated with 1 or 2 lines of therapy: the final overall survival was 10.3 months with ivosidenib and 7.5 months with placebo, but when adjusted for crossover, the median overall survival with placebo was 5.1 months. 85 Data presented at the ASCO Gastrointestinal Cancers Symposium showed that mIDH1 patients enrolled in the ClarIDHy study remaining on Ivosidenib treatment maintain a quality of life better than those on placebo treatment. 86 The analysis of 65 iCCA patients with mIDH showed a mOS and mPFS of 21.2 months and 8.3 months, respectively, following standard first-line chamotherapy. 87 mOS was significantly longer in patients with mIDH compared to those with IDH-WT (21.2 months vs 10.5 months); in patients receiving IDH inhibitor therapy, mPFS was 4.6 months with a disease control rate of 29%. 80 These observations supported the view that iCCA with mIDH represents a unique subtype of iCCA with a better OS compared to IDH-WT CCA; IDH inhibitors represent a promising therapeutic option in later lines of therapy.
Other studies have explored the targeting of the HER2 pathway in patients with advanced BTC. The analysis of 1863 CCA patients with NGS showed a high prevalence of HER2 alterations: 4.2% in iCCA. And 9.7% in eCCA. 88 Among HER2-altered patients, 23.8% of iCCA and 53.6% of eCCA patients displayed a point mutation in HER2, while 66.6% of iCCA and 41.2% of eCCA patients had HER2 copy number amplification. 81 The MyPathway trial explored the activity of pertuzumab (anti-HER2 mAb) combined with trastuzumab (anti-HER2 mAb) in 39 chemorefractory, advanced stage BTC with HER2 overexpression, HER2 amplification, or both, and observed an ORR of 23% (9/39 partial responses). 89 The pan-HER inhibitor neratinib was explored in BTC harboring a HER2 mutation and an ORR of 12% was reported. 90 The final results of the SUMMIT trial (NCT 01953926) based on the administration of neratinib to HER2 mutation-positive patients with BTC showed in the 11 iCCA-treated patients a PFS of 14 months, an OS of 5.4 months; worse outcomes were observed for tumors with co-occurring TP53 and CDKN2A alterations. 91
A recent preclinical study showed that lapatinib and lapatinib-gemcitabine combination treatment elicited a significant antitumor effect in HER2-overexpressing CCAs, including those gemcitabine-resistant, in organoid and cell line models. 92
BRAF mutations represent another potential molecular and therapeutic target of iCCAs: in fact, BRAF mutations have been observed in 3% to 5% of these tumors. Studies carried out in other tumors, particularly in melanomas, have shown that these mutations are suitable therapeutic targets and tumors bearing these mutations can be treated using BRAF inhibitors, such as dabrafenib and vemurafenib. A multicenter phase II basket trial, Oncology Agnostic Research (ROAR), explored the therapeutic impact of a BRAF inhibitor for patients with BRAFV600E-mutated rare cancer: this trial included also 43 patients with advanced BTC. 93 The results of this trial showed that the combination of a BRAF inhibitor (dabrafenib) with a MEK inhibitor (trametinib) resulted in an ORR of 47%. 93
BRCA mutations represent another potential therapeutic target of CCAs. The characterization of a large cohort of patients with BTC showed that: overall, BRCA1 and BRCA2 mutations are observed in 3.6% of samples (0.6% BRCA1% and 3% BRCA2); in iCCA, BRCA2 mutations (2.7%) are more frequent than BRCA1 mutations (0.4%); in eCCA, BRCA1 (2.1%) and BRCA2 (2.6%) are similar in their mutational frequency; BRCA2 mutations are more frequently observed in patients with high mutational burden and with MSI-H.94 BRCA-mutant CCAs are sensitive to PARP inhibitors.
Integrative studies englobing the clinical, genomic, and transcriptomic profiles of tumor cells and the transcriptomic profile of tumor microenvironment have identified several tumor subtypes and have highlighted also the existence of a link between tumor molecular abnormalities and certain immune-related properties of the tumor microenvironment. Biliary tract cancers display immunogenic features, such as the expression of molecules that may inhibit the antitumor immune response, including programmed death 1 (PD-1) and its ligand (PD-L1) and cytokine T-lymphocyte-associated protein 4. (CTLA-4), in the tumor microenvironment. These observations, as well as the recent important clinical, progresses at the level of tumor immunotherapy have supported the development of clinical studies of immunotherapy in cholangiocarcinoma using immune check inhibitors. In this context, the interim results of an ongoing phase III clinical study involving the enrollment of unresectable, locally advanced, recurrent, or metastatic patients, randomized to receive either placebo + gemcitabine/cisplatin or durvalumab (anti-PDL1) + gemcitabine/cisplatin: durvalumab + gem-cis group showed increased overall survival compared to the group treated with placebo + gem-cis (24.9% vs 10.4% at 24 months). 95 The final results of this trial confirmed these results and showed also an improvement of PFS. 96 A phase II nonrandomized trial evaluated standard chemotherapy (gemcitabine and cisplatin) plus durvalumab (anti-PD-L1 antibody) with or without trelimumab (anti-CTLA-4 antibody) in chemotherapy-naïve patients with advanced BTC: 50% of the patients in the chemotherapy alone group achieved a response, compared to 72% in the chemotherapy plus durvalumab group and 70% in the chemotherapy plus durvalumab plus tremelimumab group. 97
Another study evaluated the anti-PD-1 antibody nivolumab in the treatment of patients with advanced biliary tract cancer: the objective response was observed in 21% of patients with iCCA and 40% with eCCA. 98 Two studies evaluated the anti-PD-1 antibody pembrolizumab in advanced BTCs. In the KEYNOTE-158 study, 104 patients were enrolled and in the KEYNOTE-028 study, 24 patients were enrolled.99,100 In the KEYNOTE-158 study, the ORR was 6%, with a median PFS of 2 months and a median OS of 7.4 months; in the KEYNOTE-028 study, the ORR was of 13%, with a median OS of 6.2 months.99,100
Yoon et al explored the molecular features associated with response or resistance to immunotherapy. 18 In PD-1/PD-L1 blockade-treated patients, KRAS alteration and chromosomal instability tumors were associated with resistance to immunotherapy: the majority (about 95%) of patients with these tumor features have a low mutation burden and do not show clinical benefit following PD-1/PD-L1 blockade therapy. 18 Low tumor-infiltrating lymphocyte. (TIL) cell density was associated with an immune suppressive microenvironment and a poor response to immunotherapy, whereas high intratumor TIL density was associated with a favorable response to immunotherapy. 18
Concluding Remarks
A better definition of the molecular mechanisms underlying the development of cholangiocarcinoma and the definition of molecular subtypes represent a fundamental step for the identification of new therapeutic strategies.
Footnotes
Ethical Statement
Our study did not require ethical board approval. Because did not contain human or animal trials.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.