
Abstract
It has been proposed that CRPC treatment with reduced systemic toxicity can be achieved by employing genes that express enzymes that activate pharmacological agents. In this paper, we report our study that used human adipose-derived stem cells (ADSC), rabbit CE, and human TRAIL with reduced toxicity to explore how tumor development can be suppressed in CRPC-bearing mouse models. In vitro and in vivo directional migration of ADSC.CE.sTRAIL cells toward PC3 cells was significantly stimulated.
ADSC.CE.sTRAIL showed higher suicide effects than did ADSC, ADSC.CE, or ADSC.sTRAIL under CPT-11 treatment. PC3 cells co-cultured with ADSC.CE.TRAIL showed higher cytotoxicity than did CPT-11 monotherapy, ADSC.CE, or ADSC.sTRAIL under CPT-11 treatment. ADSC.CE.sTRAIL showed higher apoptosis than did CPT-11 monotherapy, ADSC.CE, or ADSC.sTRAIL under CPT-11 treatment. In the in vivo study, ADSC.CE.sTRAIL inhibited tumor growth more than did CPT-11 monotherapy, ADSC.CE, or ADSC.sTRAIL under CPT-11 treatment. The evidence suggests that patients’ own ADSC could be used in clinical trials for CRPC treatment based on therapeutic stem cells that express CE and TRAIL complex genes.
Introduction
In advanced prostate cancer, hormonal therapy causes remarkable improvement in bone pain and a decrease in prostate-specific antigen level. However, most cancers eventually become castration-resistant prostate cancer (CRPC) at approximately 12–18 months after castration. After diagnosis of CRPC, life is usually limited to less than two years, and palliative therapies have been the mainstay. 1
Although there have been many attempts with chemotherapies to treat CRPC, the outcomes were disappointing. If a high dose of chemotherapy drugs can be applied, the optimal antitumor effect is expected; however, their severe toxicity limits their use.
Pharmacological agents used in chemotherapy have elevated systemic toxicity. It is possible to diminish this toxicity by employing pro-drug enzymes to convert substrates with reduced toxicity. Nevertheless, therapeutic gene hypo-expression is the major reason that benefits for treatment have been minor, so encouraging empirical outcomes have not amounted to any notable patient cures. For gene-therapy technology to be effective, the therapeutic product must be introduced into the right target cells. Hence, a cell-based method of delivery with good potential is the insertion of target genes into different types of stem cells.2,3
The pro-drug CPT-11 (Irinotecan), a derivative of the natural alkaloid camptothecin, is one of the most active drugs in the treatment of colorectal cancer, with promising activity against other solid tumors. 4 Irinotecan is activated by human-liver carboxylesterase (CE) to generate SN-38 (7-ethyl-10-hydroxy-camptothecin), a topoisomerase I inhibitor that is 100–1000 times more potent than is Irinotecan in vitro and in vivo. 4
The liver, small intestine, kidney, and lung all contain CE, with the first two being particularly rich in these enzymes, which play a key role in the first-pass metabolic hydrolysis of substrate pharmacological agents. Critical drug-metabolism pathways in humans are human CE 1 and human CE 2. Although the liver is associated with expression of both these CE, human CE 1 is much more available than is human CE 2. 5
However, CE is not present in human blood, so CPT-11 conversion in human plasma is minimal. Therapeutic effectiveness can be increased by combining CPT-11 with CE from rabbit liver, which has 100 times greater efficiency than does human CE. 6 Hence, rabbit-liver CE 1 was chosen for our purposes in this paper. Gene products could be delivered to tumor sites in a targeted manner via genetic engineering, thus enabling tumor eradication. Their genetic modification could be exploited to deliver gene products to specific tumor sites to eliminate tumors.
Tumor necrosis (TNF)-related apoptosis-inducing ligand (TRAIL) has good potential as a cancer treatment tool because it can selectively trigger apoptosis in different tumor cells, while leaving healthy cells and tissues intact.7,8
In multicellular organisms, homeostasis depends significantly on the biological process with genetic regulation known as apoptosis. Apoptosis-triggering proteins, unfavorable growth environment, radiation, and chemicals are among the factors that activate apoptosis. For proteins that can trigger apoptosis, ligand-type cytokine molecules of the TNF family are well known. As TRAIL receptors are widely expressed, TRAIL can target most tissues and types of cells. TRAIL is a promising cancer therapeutic tool because it selectively induces apoptosis in tumor cells both in vitro and in vivo. 8
The expression of TRAIL takes the form of a type 2 transmembrane protein, which gives rise to a homotrimer that binds three receptors that are found at the interface between two of its subunits. 9 It is believed that the best biological activity is achieved when the trimeric ligand contains a cysteine-bound zinc atom.9,10 One study reported that marked antitumor effect with no systemic toxicity was accomplished when recombinant soluble TRAIL was administered to athymic or severe combined immunodeficient mouse models that carried human tumor xenografts. 10
The detection of receptors and apoptosis-related activity is critically dependent on the homotrimer formed by soluble TRAIL.8,9 The secretable trimeric protein TRAIL forms (sTRAIL) were generated, and apoptosis-related activity was increased over that of the known TRIAL protein by the development of a secretion signal, a domain with trimer formation, and the human TRAIL gene (amino acids 114-281) that encoded the apoptogenic receptor-binding moiety. 11
Recently, evidence has been produced that TRAIL-triggered apoptosis in prostate and breast cancer, colon carcinoma, and glioma cell lines was intensified by the natural alkaloid camptothecin derivative called CPT-11 (Irinotecan).10,12,13 One study reported that a prostate-cancer tumor was brought under control by administering TRAIL and CPT-11 jointly, which regulated proteins belonging to the Bcl-2 family and markedly activated caspases. 13
Combined treatment with TRAIL and CPT-11 can potentiate the antitumor effect without increasing the concentration of CPT-11, thus producing far less toxicity. The generation of a sTRAIL-overexpressing stem cell allows sTRAIL to be released at high concentrations from stem cells for a long time.
For gene therapy technology to be effective, therapeutic agents must be administered to the right target cells. Stem cells act as delivery vehicles that can disseminate therapeutic gene products specifically to invasive tumor cells, so the use of these therapeutic stem cells is highly attractive. A cell-based method of delivery that shows great promise is the insertion of target genes into different types of stem cells.
The reason that gene insertion into stem cells is a promising approach to cell-based delivery is that stem cells can migrate toward tumor sites.2,3,14,15 Stem cells may move to and become embedded into the tumor mass because of the requirement for the development of tumor stroma, which includes tissue remodeling whereby stem cells proliferate abundantly as the tumor expands. The tumor mass may consist largely of stem cells with external administration.3,16–18
A new strategy for possibly surpassing the limitation of chemotherapy application has been recently proposed based on the identification of the innate tumor-trophic properties displayed by stem cells. As stem cells can disseminate therapeutic gene products specifically to invasive tumor cells, the use of these therapeutic stem cells is highly attractive.
Adipose-derived stem cells (ADSC) can be the best option for clinical application. Among the advantages of these cells are straightforward acquisition without ethical considerations about the source of stem cells, fast expansion to the necessary quantity, and amenability to gene manipulation ex vivo. 19 As obtaining ADSC needs anesthesia, and storage of ADSC is difficult, the generation of immortalized stem cells can be a good solution. As hTERT does not induce malignancy if ADSC is modified with hTERT, immortalized hTERT-ADSC can be a vehicle for the transduction of therapeutic genes. 19
As stem cells can be easily transduced with viral vectors, the durable expression of therapeutic genes in stem cells is possible, which can facilitate the administration of therapeutic genes with cancer specificity for tumor eradication. Therapeutic gene-modified ADSCs are killed by the highly toxic drug activated by therapeutic genes as wells as are tumor cells. Our purpose in this work is to find out whether tumor growth can be more efficiently suppressed in CRPC-bearing mouse models by reducing toxicity with rabbit CE and human sTRAIL-overexpressing human TERT immortalized human ADSC (hTERT-ADSC.CE.sTRAIL).
Materials and Methods
This study has been approved by Animal Experimental Center (2019-4). Much of the method is based on our previous work. 20 The reporting of this study conforms to ARRIVE 2.0 guidelines. 21
Cell Culture
We purchased an ASC52TELO, hTERT-ADSC (ATCC
We cultured a PC3 prostate-cancer cell line of mouse (Korean cell line bank, Seoul, Korea) in DMEM supplement with 2 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, and 10% heat-inactivated FBS (GibcoBRL). All cells were propagated in 5% CO2, 95% air at 37 °C in a humidified incubator and routinely passaged using trypsinization.
Generation of hTERT-ADSC.CE.sTRAIL Lines
We generated recombinant lentivirus from the CLV-Ubic
Analysis of hTERT-ADSC.CE
RT-PCR
We extracted total RNA from selected cell clones using the “TRIzol reagent” (GIBCO-BRL, Life Technologies, Grand Island, NY). We did reverse transcription using total RNA and did random primer RT-PCR using the following primers:
[CE] Forward: 5’- TGCTGGGCTATCCACTCTCT −3’; Reverse: 5’- CTCCAGCATCTCTGTGGTGA-3’. [sTRAIL] Forward: 5’- GGATGAAGCAGATCGAGGAC-3’; Reverse: 5’-GCCTCTGGTGCCAGTAATGT-3’ (Table 1).
Sequence of PCR Primers.

β-Actin controls confirmed equal protein loading. The amplification program was as follows: a denaturation step of 3 minutes, followed by 35 cycles at 95 °C for 1 minute, 63 °C for 1 minute, and 72 °C for 1 minute. We examined amplification products by electrophoresis in 1.2% agrose 1× TAE gels and ethidium bromide staining.
Western Blot Analysis
Using RIPA lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA), we extracted the entire proteins from the cell lysates. Then, we did sodium dodecyl sulfate–polyacrylamide gel electrophoresis to isolate the proteins, which were then transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) for Western blotting. We used 5% skim milk to block the membranes, which were then incubated with specific primary antibodies against CE (1:1,000, abcam, Cambridge, UK), TRAIL (1:1,000, abcam), β-actin (1:3,000, Santa Cruz Biotechnology, Dallas, TX, USA), stem-cell markers including Oct4 (1:1000, Thermo Fisher), Nanog (1:1000, Thermo Fisher), and Sox2 (1:1000, Thermo Fisher) after blocking. Later, we used peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) as secondary antibodies to the membrane. We analyzed bands following their visualization using enhanced chemiluminescence reagent (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK).
Flow Cytometry Analysis
We used a Cyflow Cube 8 FACS instrument (SysmexPartec, Görlitz, Germany) and analyzed data using standard FSC Express software (De Novo Software, Los Angeles, CA, USA). We subjected hTERT-ADSC, hTERT-ADSC.CE, hTERT-ADSC.sTRAIL, and hTERT-ADSC.sTRAIL cells to flow cytometry analysis using anti- CD29 (1:50, Origene Technologies, Rockville, MD, USA), CD 34 (1:50, Invitrogen, Carlsbad, CA, USA), CD 45 (1:50, LDBio, Seattle, WA, USA), CD 90 (1:50, Origene Technologies), and CD 105 (1:50, Origene Technologies) antibody. We used a two-color flow cytometry system (BD FACS Canto II; BD, Franklin Lakes, NJ, USA) to examine the immunostained cells. By comparing the results with corresponding negative controls, we calculated the percentage of stained cells.
Migration Ability of hTERT-ADSCs.CE.sTRAIL Toward Prostate Cancer
Cell Invasion Assays
We evaluated the tropism of hTERT-ADSC, hTERT-ADSC.CE, hTERT-ADSC.sTRAIL, and hTERT-ADSC.sTRAIL for tumor cells in transwell cell-culture chambers with Matrigel coating and a pore size of 8 μm (Merck Millipore, Billerica, MA, USA). Experiment commencement was preceded at 24 hr by loading of medium or 1.5 × 104 WPMY-1, human prostatic myofibroblast cell-line cells (ATCC, Manassas, VA, USA) in the lower wells of the plates with 24 wells. The ECMatrix-coated inserts with a pore size of 8 Am received the hTERT-ADSC, hTERT-ADSC.CE, hTERT-ADSC.sTRAIL, or hTERT-ADSC.sTRAIL (105 cells) in medium without serum. We used PBS to wash the cell-containing lower wells, which were subsequently supplemented with medium without serum, amassed for the migration assay, and subjected to 48-hr incubation. The next step was removal of non-invading cells and ECMatrix from within the inserts, followed by 20-minute staining of the cells and microscopic imaging of the invading cells. Assessment of the results involved direct counting of the number of migrated cells in five fields and mean calculation.
In Vivo Migration Studies
We conducted all procedures in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (2001) and also in accordance with guide for the care and use of laboratory animals. 22 They were approved by the Institutional Animal Care and Use Committee of the Hospital. We purchased adult male nude mice, each weighing 20–25 g (OrientBio, Seognam, Korea). The mice were kept in temperature-regulated conditions, with an alternating 12-hr cycle of night and day, and were provided with standard food and unlimited water. We used isoflurane (BKPharm, Goyang, Korea) to anesthetize the mice before we used a 30-gauge Hamilton syringe for subcutaneous injection of 1.0 × 106 PC3 cells in 100 μl PBS in the animals’ flanks. Two weeks following PC3 injection, we used isoflurane again to anesthetize the mice. We then used a 100-μm syringe with 26G needle for the injection of PBS or 1 × 106 hTERT.ADSC.CE.TRAIL cells combined with 100 μl saline into the animals’ left ventricles. After 2 weeks, they were sacrificed and induced prostate cancer at the flank was removed. We did real-time PCR for induced tumors at the flank by PBS, hTERT-ADSC, or hTERT-ADSC.CE.sTRAIL via intracardiac injection to see whether tumors demonstrated the presence of GFP, rabbit CE, and sTRAIL in induced tumors from mice for GFP, CE, sTRAIL, and β-actin.
We did quantitative real-time PCR (qRT-PCR) analysis using Maxima STBR Green/ROX qPCR Master Mix (Thermo Fisher). We did qRT-PCR using the PIKOREAL 96 (Thermo Fisher) under cycling conditions: 95 °C for 15 seconds (denaturation), 55 °C for 30 seconds (annealing), and 72 °C for 60 seconds (extension) for 45 cycles. We then calculated the gene expression level, normalized to β-actin, using the 2-ΔΔ formula with reference to the ADSC.CE.sTRAIL. Primers were as in the Table 1.
Chemoattractant Ligand and Receptor Validation
We evaluated the presence of the ligands, stem-cell factor (SCF), stromal cell-derived factor 1 (SDF-1), and vascular endothelial growth factor (VEGF) in PC3 cells and their corresponding receptors, c-kit, chemokine receptor 4 (CXCR4), VEGF receptor (VEGFR)-1, VEGFR2, and VEGFR3 in hTERT.ADSC, hTERT.ADSC.CE, and hTERT.ADSC.CE.sTRAIL cells. Table 1 indicates the chemoattractant ligands and receptors (ie, sense and anti-sense primers) as well as the estimated sizes of the yields from the RT-PCR reaction.
Experiments In Vitro
Suicide Effect of hTERT.Ce.sTRAIL/CPT-11 System
We assessed suicide to CPT-11 for hTERT-ADSC, hTERT-ADSC.CE, hTERT-ADSC.sTRAIL, or hTERT-ADSC.sTRAIL cells. We plated cells (5 × 103 cells/well) in 96-well plates (Falcon, Becton-Dickinson Co., Franklin Lakes, NJ). After 24 hr, we treated cells with CPT-11 (Sigma-Aldrich) with the last concentrations of 0–5 μM. After 72-hr incubation at 37 °C, we assessed the cells for viability. All the experiments were done in quadruplicate. We assessed cell viability via a modified MTT assay (Promega, Madison, WI, USA), in which mitochondrial dehydrogenase was the basis for converting MTT tetrazolium salt to a formazan product. After addition of 10-μl MTT solution to every well, we did a 4-hr incubation at 37 °C. We used dimethyl sulfoxide (Sigma-Aldrich) for color extraction at 37 °C for 20 minutes. We used a microplate reader (Infinite F50; Tecan, Männedorf, Switzerland) to measure the reaction absorbance at 570 nm and thus quantify the formazan product. Cell viability was expressed as the mean ± standard error (SE) as a percentage of the control viability.
Cytotoxicity of hTERT-ADSC.Ce.sTRAIL to Pc3 Cells
In Vivo Inhibition of Tumor Growth in ADSC.CE.sTRAIL
We followed our previous animal model during in vivo experiments.20,23 We anesthetized the mice with isoflurane (BKPharm) with 3%-4% for induction and 1%-3% for maintenance, after which we used a 30-gauge Hamilton syringe for subcutaneous injection of 1.0 × 106 PC3 cells in 100 μl PBS into the animals’ flanks. At 1 and 2 weeks after intracardiac injection of PBS (n = 5), hTERT-ADSC (n = 5), hTERT-ADSC.CE (n = 5), hTERT-ADSC.sTRAIL (n = 5) or hTERT-ADSC.CE.sTRAIL (n = 5), we treated the animals with 1.7 mg/kg/day of CPT-11 diluted in 100 μl PBS, which was administered through intraperitoneal injection in two rounds of five sequential days with a two-day intermission. We used calipers to measure tumor volumes at days 0 and 14 post-injection. The formula for volume calculation was volume = length × width
To evaluate the effect of high concentration compared with low concentration of CPT-11, at weeks 1 and 2 after intracardiac injection of hTERT-ADSC.CE (n = 5), hTERT-ADSC.sTRAIL (n = 5), or hTERT-ADSC.CE.sTRAIL (n = 5), animals were treated with 13.5 mg/kg/day of CPT-11 diluted in 100 μl PBS i.p. in two rounds of 5 consecutive days with a break of 2 days. On days 0 and 14 after injection, we measured the tumor volumes. The CPT concentration was assessed by preliminary experiments on the tumor percent volume administered at the first administration and the second week. We evaluated results as the mean of tumor volume ± SE. On 14 days after injection, we sacrificed the mice and removed induced tumors. Euthanasia was performed using inhalant euthanasia agent of carbon dioxide (CO2).
Imuunohistochemistry
We did the immunohistopathological examination of removed tumors for TRAIL to detect if delivered TRAIL was present and involved in anticancer activity. We used a TRAIL antibody (1:100 ADI-AAP-470; Enzo Life Sciences, Farmingdale, NY) and a DAB Detection IHC Kit (abcam). We did the TRAIL assay for tumor tissues that were removed after 2 weeks treatment of hTERT-ADSC.sTRAIL and hTERT-ADSC.sTRAIL in combination with CPT-11. We visualized the stained sections using a microscope (Olympus, Tokyo, Japan), using low-power (X100) or high-power (X400) fields of slides.
For the detection of apoptosis, we did a terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL) assay. We used a TdT apoptosis detection kit (Takara BIO, Mountain View, CA) and a DAB Detection IHC Kit (abcam). We did the TUNEL assay for tumor tissues, which were removed after 2 weeks of treatment with hTERT-ADSC.sTRAIL and hTERT-ADSC.sTRAIL in combination with CPT-11. We visualized the stained sections using a microscope (Olympus), using low-power (X100) or high-power (X400) fields of slides.
Statistics
To find out whether the results from the evaluation of cell viability and tumor volume differed significantly, we did a two-way ANOVA and post hoc Tukey test. Data are presented as means ± SE, and p < .05 was considered to be statistically significant.
Results
Generation of hTERT-ADSC
An ASC52TELO, hTERT-ADSC (ATCC
Gene Expression of TRAIL from hTERT-ADSC.CE.sTRAIL
The hTERT-ADSC.CE.sTRAIL cells were produced by lentiviral transduction of the CE.sTRAIL gene using CLV-Ubic blastacicin/ CE.sTRAIL vector (Figure 1A). We confirmed the expression of the CE.sTRAIL transcript from hTERT-ADSC.CE.sTRAIL by RT-PCR. CE.sTRAIL transcripts were demonstrated only in hTERT-ADSC.CE.sTRAIL, but not in hTERT-ADSC cells (Figure 1B). We confirmed the expression of CE.sTRAIL protein from hTERT-ADSC.CE.sTRAIL by Western blot. CE.sTRAIL protein was demonstrated only in hTERT-ADSC.CE.sTRAIL, but not in hTERT-ADSC cells (Figure 1C). Phase-contrast microscopy of hTERT-ADSC, hTERT-ADSC.CE, ADSC.sTRAIL, and hTERT-ADSC.CE.sTRAIL cells were spindle shaped (Figure 1D).

CE, sTRAIL, CE.sTRAIL overexpressing hTERT immortalized human adipose stem-cell line. A, hTERT-hADSC, hTERT-hADSC.sTRAIL, and hTERT-hADSC.CE.sTRAIL were generated by a lentiviral transduction of GFP, CE, sTRAIL, CE.sTRAIL gene using a CLV-Ubic vector. B, C, We confirmed the expression of GFP, CE, sTRAIL, CE.sTRAIL transcript or protein by RT-PCR or Western blot. D, Phase-contrast microscopy of hTERT-hADSC.GFP, hTERT-hADSC.sTRAIL, and hTERT-hADSC.CE.sTRAIL line.
Phenotype of hTERT-ADSC.CE.sTRAIL
The hTERT-ADSC, hTERT-ADSC.CE, ADSC.sTRAIL, and hTERT-ADSC.CE.sTRAIL cells express cell-type-specific mesenchymal stem-cell markers for CD29, CD90, and CD105, but not for hematopoietic stem cells CD34 or CD45 (Figure 2A). Stem-cell markers, including Oct4, Nanog, and Sox2, were expressed in the hTERT-ADSC, hTERT-ADSC.CE, ADSC.sTRAIL, and hTERT-ADSC.CE.sTRAIL cell line. The maintenance of human MSCs was shown (Figure 2B).
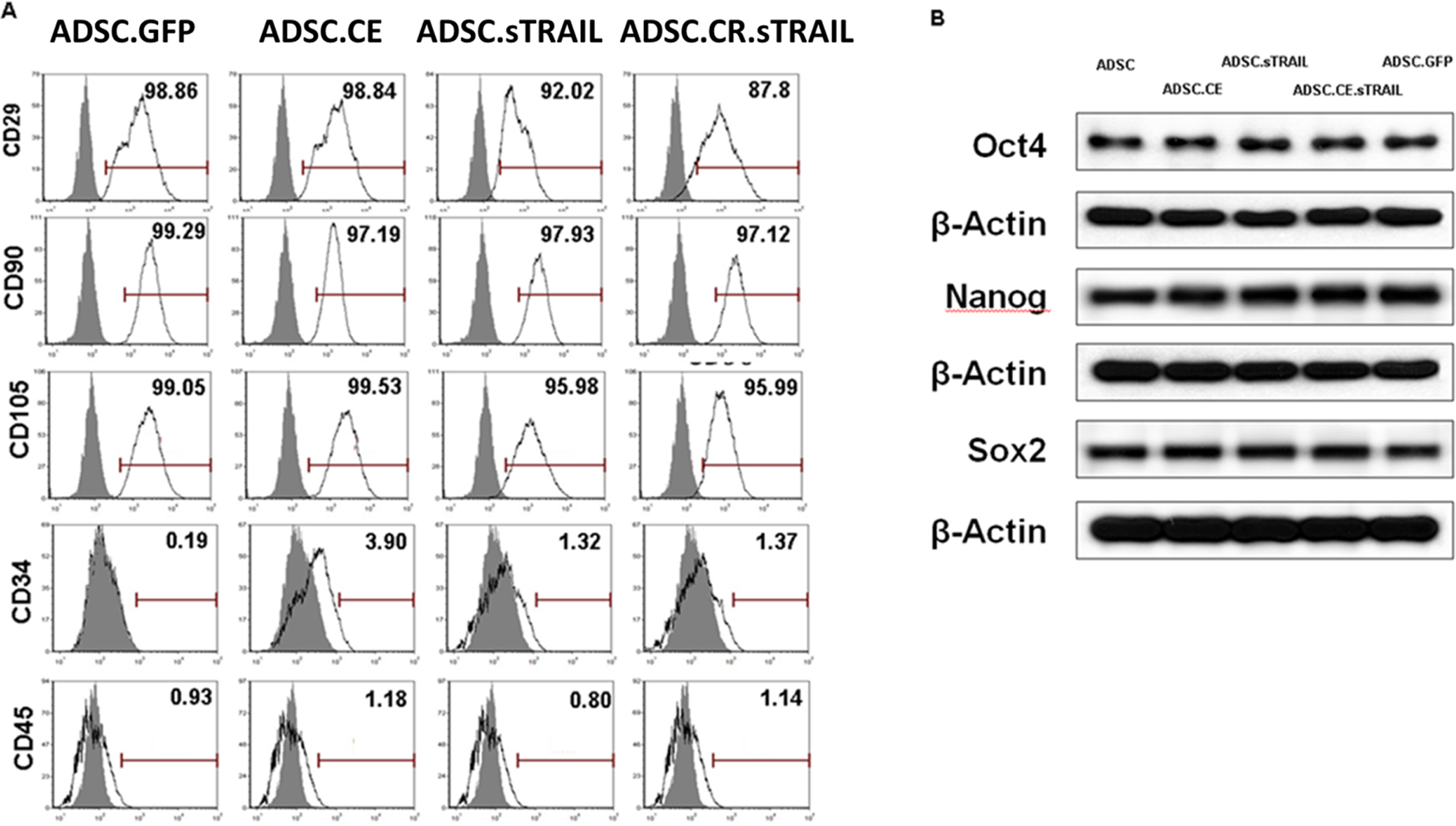
Cell markers of hTERT-hADSC.GFP, hTERT-hADSC.GFP.CE, hTERT-hADSC.GFP.sTRAIL, and hTERT-hADSC.GFP.CE.sTRAIL cells. They express cell-type-specific markers for mesenchymal stem cells CD29, CD90, and CD105, but do not express cell-type markers for hematopoietic stem cells CD34 or CD45. They express cell-type-specific markers for stem cells Oct4, Nanog, and Sox2.
Migration of hTERT-ADSC Cells Toward Cancer Cells
Invasion Study
Whereas few hTERT-ADSC, hTERT-ADSC.CE, hTERT-ADSC.sTRAIL, or hTERT-ADSC.CE.sTRAIL cells migrated toward media or normal prostate-cancer cells (WPMY-1) was stimulated, the directional migration of hTERT-ADSC, hTERT-ADSC.CE, hTERT-ADSC.sTRAIL, and hTERT-ADSC.CE.sTRAIL cells toward PC3 cells exhibited marked in vitro stimulation by human prostate cancer cells (PC3). On the other hand, migration ability was unaffected by transgenic expression in CE, TRAIL, or and CE.TRAIL or by treatment in vitro at CPT-11 5 μM for 72 hr (Figure 3A and B).

The hTERT-ADSC, hTERThADSC.CE, and hTERT-ADSC.CE.sTRAIL cell lines target prostate cancer. A, B, Invasion study showed the migration of hTERT-ADSC or hTERT-ADSC.CE to prostate cancer. C, Intracardiac-delivered hTERT-hADSC.CE.sTRAIL targets prostate cancer. Real-time PCR demonstrated the presence of GFP, rabbit CE, and human sTRAIL in resected tumors from mice. D, The expression of chemoattractant factors. We confirmed the presence of the ligands SCF, SDF-1, and VEGF in TRAMPC2 cells and of their corresponding receptors, c-kit, CXCR4, VEGFR1, VEGFR2, and VEGFR3, in hTERT-ADSC, hTERThADSC.CE, and hTERT-ADSC.CE.sTRAIL cells.
In Vivo Migration Study
Real-time PCR demonstrated the presence of GFP, rabbit CE, and human sTRAIL from the removed tissue of induced prostate cancer in mice. The mRNA level of GFP (fold) was not detectable in the PBS treatment group. However, it was shown in ADSC (1.0 ± 0.1). The mRNA level of the CE-treated group (3.7 ± 1.1) was higher than that in the ADSC treatment group (1.0 ± 0.1) (Figure 3C) (p < .05). The mRNA level of the sTRAIL-treated group (2.7 ± 0.7) was higher than that in the ADSC treatment group (1.0 ± 0.1) (Figure 3C) (p < .05). As GFP, rabbit CE, or human sTRAIL genes do not exist in mouse tissue, the presence of rabbit GFP, CE, or human sTRAIL genes demonstrated the selective migration of hTERT-hADSC.CE.sTRAIL toward induced prostate cancer of mice. The intracardiac delivery of hTERT-hADSC.CE.sTRAIL toward targeted prostate cancer was confirmed after systemic injection.
Confirmation of Chemoattractant Ligands and Receptors
Using reverse transcription PCR, we confirmed the presence of the ligands SCF, SDF-1, and VEGF in PC3 prostate-cancer cells and of their corresponding receptors c-kit, CXCR4, VEGFR1, VEGFR2, and VEGFR3 in hTERT-ADSC, hTERT-ADSC.CE,s hTERT-ADSC.sTRAIL, and hTERT-ADSC.CE.sTRAIL cells (Figure 3D).
In Vitro Selective Cytotoxic Effects of hTERT-ADSC.CE.TRAIL
In Vitro Suicide Effect of hTERT-ADSC.Ce.sTRAIL
To find the concentration of CPT-11 to use in these assays, we experimented with the response of hTERT-ADSC, hTERT-ADSC, hTERT-ADSC.CE, hTERT-ADSC.TRAIL, and hTERT-ADSC.CE.TRAIL cell lines to CPT-11 using the cell viability assay. By comparison to the hTERT-ADSC group (106.8 ± 1.5), there was a decrease in the proportion (%) of cell viability of stem cells in hTERT-ADSC.CE (100.1 ± 4.8), hTERT-ADSC.sTRAIL (100.6 ± 1.8), and hTERT-ADSC.CE.sTRAIL (90.3 ± 3.0) at 0.05 μM (p < .05), according to in vitro analysis. Likewise, in comparison to the hTERT-ADSC group (80.3 ± 2.0), there was a decrease in the proportion of cell viability in hTERT-ADSC.CE (69.3 ± 3.8), hTERT- ADSC.sTRAIL (84.4 ± 1.2), and hTERT-ADSC.CE.sTRAIL (46.8 ± 2.9) at 5 μM (p < .05). The cell viability of hTERT-hADSC.CE.sTRAIL decreased according to the concentration-dependent pattern of CPT-11 at more than the concentration of 0.05 μM CPT-11. All of the hTERT-ADSC.CE, hTERT-ADSC.TRAIL, and hTERT-ADSC.CE.TRAIL cell lines showed suicide effects with the final concentrations of 5 μM. For the effective in vitro suicide effect of hTERT-hADSC.CE.sTRAIL under the treatment with CPT-11, the concentration of CPT-11 at more than 0.05 μM was necessary. The IC50 value, defined as the dose required for 50% cytotoxicity, for CE-transduced hTERT-ADSC.CE.TRAIL cells was approximately 5 μM at 72 hr (Figure 4A).

In vitro effect of hTERT-ADSC.CE under the treatment with CPT-11. A, The suicide effect. At more than 0.05 μM CPT-11, cell viability of ADSC.CE was lower than that of ADSC. B, The cytotoxicity of ADSC.CE.sTRAIL toward PC3 at co-cultured ratios of 0.05 under 5 μM CPT-11. Cell viability of PC3 decreased significantly under the treatment with both CPT-11 and ADSC.CE compared to that of CPT-11 monotherapy. C, Flow cytometry for PI/Annexin V staining showed that the percentage of Annexin V–positive apoptotic PC3 cells increased in the presence of ADSC.CE under CPT-11 more than with CPT-11 monotherapy. D, The percentage of apoptosis significantly increased in the hTERT-ADSC.CE.sTRAIL-treated group more than in the hTERT-ADSC or CPT-11 monotherapy (p < .05). ADCS = hTERT-hADSC; ADSC.CE.sTRAIL = CE.sTRAIL overexpressing hTERT immortalized human adipose stem cells. *p < .05.
In Vitro Cytotoxicity of hTERT-ADSC.Ce.sTRAIL
When CPT-11 was present, hTERT-ADSC.CE.sTRAIL provided mediation for selective cytotoxicity for PC3 cells at 5 μM for 3 days. In comparison to the hTERT-ADSC group (100.0 ± 1.0), co-culturing of PC3 cells with hTERT-ADSC in the presence of CPT-11 reduced the proportion of PC3 cell viability (%) in hTERT-ADSC.CE (74.1 ± 2.0), hTERT-ADSC.sTRAIL (97.8 ± 0.4), and hTERT-ADSC.CE.TRAIL (49.2 ± 0.3). Even for a ratio as low as 0.1, hTERT-ADSC exhibited potent selective cytotoxicity against PC3 cells in the presence of CPT-11 (Figure 4B).
In Vitro Apoptotic Effect of hTERT-ADSC.Ce.sTRAIL
For the hTERT-ADSC.CE, hTERT-ADSC.TRAIL, and hTERT-ADSC.CE.TRAIL cells, apoptosis was induced in vitro at 5 μM for 72 hr (Figure 4C). There was a marked rise in the proportion of apoptotic prostate-cancer cells positive for Annexin V in the presence of hTERT-ADSC.CE.TRAIL (73.2 ± 7.9), hTERT-ADSC.CE (39.7 ± 4.8), and hTERT-ADSC. sTRAIL (37.2 ± 2.1) with CPT-11 compared with hTERT-ADSC only (16.5 ± 1.0) (Figure 4C). The percentage of apoptosis significantly increased more in hTERT-ADSC.CE.sTRAIL cells than in hTERT-ADSC (p < .05) (Figure 4D).
As the drug concentrations were elevated, PC3 cell growth was progressively suppressed exclusively because of the presence of CPT-11. Hence, it can be deduced that, to some extent, PC3 cells express endogenous human CE that can convert CPT-11 to SN-38.
In Vivo Tumor Growth Inhibition by hTERT-ADSC.CE.sTRAIL
Systemic administration in combination with CPT-11 reduced prostate-cancer volume. At 2 weeks after CPT-11 treatment, the percentage volume of prostate cancer was 4195.0 ± 763.4 (PBS), 1442.3 ± 435.0 (CPT-11 1.7 mg/kg/day), 579.2 ± 376.7 (CPT-11 6.8 mg/kg/day), and 96.4 ± 14.9 (CPT-11 13.5 mg/kg/day). The inhibition of tumor growth was significantly CPT-11 dose-dependent (Figure 5C).

Treatment with hTERT-ADSC.CE.sTRAIL combined with CPT-11 has a significant therapeutic effect in vivo. A, Schematic summary of the treatment. B, Illustration of the induction of prostate cancer using PC3, systemic injection of hTERT-hADSC cells, and migration of the gene-modified stem cells toward the prostate cancer. Blue, PC3; red, hTERT hTERT-ADSC cells. C, Administration of CPT-11 reduced prostate-cancer volume of mice concentration dependently. *p < .05 CPT-11 0 mg versus CPT-11 1.7 mg; #p < .05 CPT-11 1.7 mg versus CPT-11 13.5 mg. D, Administration of hTERT-ADSC.CE.sTRAIL reduced prostate-cancer volume of mice more than did ADSC.sTRAIL or ADSC.CE in combination with CPT-11. ADCS = hTERT-hADSC; ADSC.CE.sTRAIL = CE.sTRAIL overexpressing hTERT immortalized human adipose stem cells.
At 2 weeks after systemic administration in combination with increased CPT-11 concentration, the percentage volumes of prostate cancer were 3708.1 ± 1154.1 (PBS), 1272.4 ± 94.8 (ADSC.sTRAIL), 1052.4 ± 140.29 (1.7 mg CPT-11/kg/day), 892.3 ± 418.0 (ADSC.sTRAIL + 1.7 m CPT-11/kg/day), 540.0 ± 201.4 (ADSC.CE + 1.7 mg CPT-11/kg/day), 471.5 ± 23.2 (ADSC.CE.sTRAIL + 1.7 mg CPT-11/kg/day), 140.2 ± 15.6 (13.5 mg/kg/day CPT-11 monotherapy), 86.7 ± 4.2 (hTERT-ADSC.sTRAIL + 13.5 mg CPT-11/kg/day), 71.2 ± 4.1 (hTERT-ADSC.CE + 13.5 mg CPT-11/kg/day), 58.1 ± 14.9 (hTERT-ADSC.CE.sTRAIL + 13.5mg CPT-11/kg/day) compared with a baseline prostate-cancer volume of 0 weeks (Figure 5D). The inhibitory effects of hTERT-ADSC.CE.sTRAIL combined with a 13.5 mg/kg/day CPT-11 concentration were better than those of CPT-11 monotherapy, hTERT-ADSC.sTRAIL, or hTERT-ADSC.CE and were dose dependent.
Hematoxylin and Eosin staining of PC3 cell transplanted tumors indicated that PC3 cells triggered the onset of prostate cancer 14 days after they were injected into the mice's flanks (Figure 6A). The tumor was well demarcated from dermal tissue and was composed of closely packed cancer cells. The neoplastic cells remaining in the peripheral lesion were shrunken and had distinct borders. Geographic necrosis was found (Figure 6A to E). Immunohistochemical staining of the removed tumor showed anti-TRAIL positive cells after hTERT-ADSC.sTRAIL or hTERT-ADSC.CE.sTRAIL combined with CPT-11 treatment (Figure 6F and G), and the hyperchromatic nuclei were centrally located with condensed eosinophilic cytoplasm, which had apoptotic bodies (Figure 6H to K).

Immunohistochemistry of removed prostate cancer. A-E, H&E staining of induced prostate cancer at 2 weeks after the implantation of PC3 cells in the flank of mice. A, Tumors in PC3 transplanted mice (X100). Inset: X400. The tumor is well demarcated from dermal tissue and is composed of closely packed cancer cells. Inset: X400. B, Tumors in PC3 transplanted mice after 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. C, Tumors in PC3 transplanted mice after hTERT-ADSC.sTRAIL and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. D, Tumors in PC3 transplanted mice after hTERT-ADSC.CE and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. E, Tumors in PC3 transplanted mice after hTERT-ADSC.CE.sTRAIL and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. Geographic necrosis was found. The neoplastic cells remaining in the peripheral lesion are shrunken and have distinct borders. F, G, TRAIL immunohistochemical staining of tumors after treatment showed anti-TRAIL positive cells. F, Tumors in PC3-transplanted mice after hTERT-ADSC.sTRAIL and 13.5 mg/kg/day CPT-11 and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. G, Tumors in PC3-transplanted mice after hTERT-ADSC.CE.sTRAIL combined with 13.5 mg/kg/day CPT-11 systemic treatment (X100), Inset: X400. H-K, TUNEL immunohistochemical staining of tumors after treatment showed anti-TUNEL positive cells. H, Tumors in PC3-transplanted mice after hTERT-ADSC.sTRAIL and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. I, Tumors in PC3-transplanted mice after hTERT-ADSC.sTRAIL. H, Tumors in PC3-transplanted mice after hTERT-ADSC.sTRAIL and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. J, Tumors in PC3-transplanted mice after hTERT-ADSC.sTRAIL and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. K, Tumors in PC3-transplanted mice after hTERT-ADSC.CE.sTRAIL and 13.5 mg/kg/day CPT-11 systemic treatment (X100). Inset: X400. The hyperchromatic nuclei are centrally located with condensed eosinophilic cytoplasm, which has apoptotic bodies. Inset: Apoptotic cells having apoptotic bodies. Bar. 500 μm.
Discussion
The pro-drug CPT-11 is one of the most active in the treatment of solid tumors (2). Irinotecan, activated by CE to generate SN-38, is more potent than CPT-11. The conversion level of CPT-11 in human plasma is very low. We did potent rabbit-liver CE gene modification to eliminate the tumors. The prostate-cancer tumor was brought under control by administering TRAIL and CPT-11 jointly, which regulated proteins belonging to the Bcl-2 family and markedly activated caspases. 13 Gene therapy can be safely delivered by engineering ADSC to undertake expression of a suicide gene/pro-drug activating enzyme system and apoptosis gene, since they can eradicate not only tumor cells via a bystander effect and apoptosis, but also ADSC themselves in vivo.
In this study, we generated human ADSC over-expressing CE and sTRAIL using immortalized human hTERT-ADSC. CE, TRAIL transcript, and protein was demonstrated only in hTERT-ADSC.CE.sTRAIL, but not in hTERT-ADSC cells (Figure 1). Gene modification was done successfully.
Some qualities set gene therapy apart from traditional drug treatments. One benefit is the straightforward achievement of high concentration and enduring expression of a pharmacological agent. Another benefit is the localized and specific delivery of pharmacological agents to the desired tissue. ADSC can be used as vehicles to deliver therapeutic genes.
An ASC52TELO, hTERT-ADSC (ATCC
Stem cells can migrate to and invade tumors, which makes them highly useful in cancer therapy. Stem cells administered through intravenous injection settle primarily in the pulmonary capillary bed. 24 As stem cells administered arterially do not localize mainly to the pulmonary capillary bed, 25 we administered stem cells via intracardiac injection in this study. Unlike liposome, antibody-drug conjugates, and nanoparticles, a stronger anti-tumor effect can be achieved by stem cells because they can invade tumor masses. Furthermore, the targeting of small remote metastases and invasion of malignant satellites following full extirpation of the primary tumor can be accomplished by taking advantage of such a tumor-specific tropism. Migratory properties were influenced by tumor-signaling molecules of the prostate cancer.3,16–18 It is possible that they are involved in such inherent tumor-specific migration capacity and interactions. Additionally, gene alteration provides no evidence to suggest that lentivirus transduction and transgene expression adversely affect the stem-cell properties of differentiation, immunophenotype, migration, or expression of pluripotency transcription factor.9,10,13 In this study, the directional migration of hTERT-ADSC.CE.sTRAIL cells toward PC3 cells was significantly stimulated by PC3 cells in vitro (Figure 3A and B). The migration ability was unaffected by CE.sTRAIL transgene expression and was preserved by the treatment with CPT-11 at 5 μM for 72 hr (Figure 3A and B).
In this in vivo migration experiment, real-time PCR demonstrated the presence of GFP, rabbit CE, and human sTRAIL from the removed tissues of induced prostate cancer in the flanks of mice after systemic treatment of hTERT-ADSC.CE.sTRAIL (Figure 3C). As rabbit CE, GFP, or human TRAIL do not exist in mice tissues, their presence demonstrated the selective migration of hTERT-ADSC.CE.sTRAIL toward induced prostate cancer of mice.
Migratory properties were influenced by tumor-signaling molecules of the prostate cancer. Recent studies suggest that the brain-tumor targeting of neural stem cells is mediated by chemoattractant molecules and their receptors, including SCF/c-Kit, SDF-1, CXCR4, VEGF/VEGFR-1, VEGFR2, VEGFR3, and epidermal and platelet-derived growth factors.3,16–18 Our work has demonstrated that the ligands SCF, SDF-1, and VEGF were present in PC3 cells, whereas their equivalent receptors, c-kit, CXCR4, VEGFR1, VEGFR2, and VEGFR3, were present in hTERT-ADSC.CE.sTRAIL cells (Figure 3D). It is possible that they contributed to the inherent ability of tumor-specific migration and interactions. Stem cells can direct migration toward the tumors.14,15,26,27 Therefore, we expected the migratory capacity of hTERT-ADSC.CE.sTRAIL to have an anticancer effect leading to cell death by stem-cell migration and the suicide gene in tumor cells.
To avoid de novo tumor formation from ADSC, hTERT-ADSC.CE.sTRAIL cells are removed by the highly toxic SN-38 drug as well as by the tumor cells. We suggest that the suicide efficiency can be improved by increasing the bystander-killing effect by lengthening SN-38 production or increasing local concentrations. These enzyme modifications lead to optimized pro-drug conversion to achieve a maximum level of the cytotoxic compound. In this study, we eliminated hTERT-ADSC.CE.sTRAIL in vitro under the treatment of CPT-11; these cells showed significantly higher susceptibility to CPT-11 than did the parental cells (hTERT-ADSC) (Figure 4A). The cell viability of hTERT-ADSC.CE.sTRAIL decreased according to the concentration-dependent pattern of CPT-11 at a concentration of more than 0.05 μM CPT-11. At a final concentration of 5 μM, cell viability was reduced in hTERT-ADSC.CE.sTRAIL (46.8 ± 2.9%) compared with that in the hTERT-ADSC group (80.3 ± 2.0%) (p < .05). For the effective in vitro suicide effect of hTERT-ADSC.CE.sTRAIL under the treatment with CPT-11, the concentration of CPT-11 at more than 0.05 μM was necessary.
The Mediation of selective cytotoxicity toward PC3 cells was provided by hTERT-ADSC.CE.sTRAIL in the presence of CPT-11 in vitro at 5 μM for 72 hr (Figure 4B). The hTERT-ADSC.CE.sTRAIL cells exert a strong selective cytotoxic effect over PC3 prostate-cancer cells, even if the ratio of hTERT-ADSC.CE.sTRAIL to PC3 cells was as low as 0.05. However, PC3 cell growth was suppressed by the CPT-11 monotherapy. Hence, to some extent, prostate-cancer cells express endogenous human CE that can convert CPT-11 to SN-38. In this study, hTERT-ADSC.CE.sTRAIL cells induced apoptosis in vitro at 5 μM for 72 hr (Figure 4C). The percentage of apoptosis significantly increased in hTERT-ADSC.CE.sTRAIL cells more than in CPT-11 monotherapy, hTERT-ADSC.sTRAIL, or hTERT-ADSC.CE. In this study, the inhibition of tumor growth by CPT-11 treatment was dose-dependent (Figure 5C). Compared with the baseline tumor-volume percent (100%), 1.7 mg or 6.8 mg CPT-11 treatment showed limited inhibition of tumor growth. When 13.5 mg was used, it showed 96%, which is less than the baseline tumor-volume percent.
With left ventricular injection of stem cells, they do not localize mainly to the pulmonary capillary bed (Figure 5B). In this study, systemic administration of hTERT-ADSC.CE.sTRAIL in combination with CPT-11 significantly reduced prostate-cancer volume. The inhibitory effects of ADSC.CE.TRAIL combined with 1.7 mg CPT-11 kg/day treatment were higher than those of PBS or CPT-11 monotherapy, ADSC.CE, or the ADSC.sTRAIL treatment group (p < .05) (Figure 5D). The inhibition of tumor volume was augmented by the increase of CPT-11 concentration of 13.5 mg CPT-11/kg/day. The inhibitory effects of hTERT-ADSC.sTRAIL combined with 13.5 mg/kg/day CPT-11 concentration were better than those of 1.7 mg/kg/day CPT-11. As the treatment with hTERT-ADSC.sTRAIL in combination with CPT-11 inhibited tumor growth better than did CPT-11 monotherapy, the concentration of CPT-11 can be reduced. Therefore, better efficacy with reduced toxicity of CPT-11 can be expected. We suggest that the antitumor effect by prolonged production of a higher local concentration of TRAIL and CE released from hTERT-ADSC.CE.sTRAIL and potentiated by CPT-11 will increase the therapeutic efficiency.
Immunohistochemical staining of the removed tumor showed anti-TRAIL positive cells after hTERT-ADSC.sTRAIL or hTERT-ADSC.CE.sTRAIL combined with CPT-11 treatment (Figure 6F and G) and the hyperchromatic nuclei were centrally located with condensed eosinophilic cytoplasm, which has apoptotic bodies (Figure 6H to K). The immunohistopathological examination of removed tumors for TRAIL demonstrated that delivered TRAIL was present and involved in anticancer activity. We suggest that TRAIL induced apoptosis in the prostate cancer and CPT-11 augmented TRAIL-induced apoptosis in prostate cancer.
In conclusion, human ADSC transduced with CE.sTRAIL exerts cytotoxicity on inserted human prostate-cancer cells when CPT-11 is present, both in vitro and in vivo. Such findings reinforce the idea that human ADSC overexpressing CE.sTRAIL could be useful in clinical trials for advanced prostate-cancer therapy.
Conclusions
Human ADSCs in combination with suitable transgene/prodrug CE.sTRAIL can migrate to the prostate cancer, improve cytotoxicity against tumor cells in vitro, and exert a very efficient tumor-growth inhibition in vivo in combination with prodrug CPT-11. The results we obtained further contribute to potential clinical utility of CE.sTRAIL gene-expressing ADSCs for CRPC.
Footnotes
Author Contributions
SHL and YSS conceived and designed the experiments. JHK, EJO, and CWY performed the experiments. JHK, SHL, and YSS analyzed the data. JHK, SHL, and YSS wrote the paper.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research was supported by grant from Soonchunhyang University Research Fund and the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2021R1A2C1004163).
Ethical Approval Statement
This study has been approved by Soonchunhyang University Seoul Hospital Animal Experimental Center (2019-4).