
Abstract
C-C motif chemokine ligand 28 (CCL28) has been reported to be pro-tumoral in several cancer types. However, the role of CCL28 in pancreatic ductal adenocarcinoma (PDAC) progression remains unclear. CCL28 mRNA expression in tumors from PDAC patients was found to be elevated as compared to normal pancreas. CCL28 expression was also negatively correlated with overall survival (OS) in pancreatic cancer patients. Our in vitro experiments showed that CCL28 knockdown impairs the proliferation of mouse pancreatic cancer cell line PAN02. Moreover, in both immunocompetent syngeneic mice and immunodeficient NOD-SCID mice, CCL28 deficiency significantly attenuated the growth of subcutaneous PAN02 tumors. In syngeneic mouse model, CCL28 downregulation remodeled the pancreatic tumor microenvironment by suppressing the infiltration of both regulatory T (Treg) cells, myeloid-derived suppressor cells, and activated pancreatic stellate cells, and upregulating the expression of lymphocyte cytotoxic proteins including perforin and granzyme B. In conclusion, our work demonstrates that CCL28 is a potential target for pancreatic cancer treatment and CCL28 blockade could inhibit tumor growth through both tumor-cell-intrinsic and extrinsic mechanisms.
Introduction
Pancreatic cancer is a highly malignant adenocarcinoma type, ranking in the top 4 for estimated deaths in the United States and with the poorest five-year survival rate among most diagnosed malignancies. 1 Among all pancreatic cancer subtypes, pancreatic ductal adenocarcinoma (PDAC) is the most frequent one and accounts for 85% to 90%. 2 Due to few clinical symptoms in the early stage, most pancreatic cancer patients were diagnosed with advanced disease and thus unresectable, though surgical resection is the only possibly curative treatment.3,4 Chemotherapies such as gemcitabine and a combination of fluorouracil and folinic acid (FOLFIRINOX) are often used as adjuvant therapies to improve overall survival (OS) after surgery.5-8 Nevertheless, these traditional therapies just showed modest efficiency in prolonging OS and chemotherapy always results in side effects and drug resistance. Immunotherapies like immune checkpoint blockade targeting cytotoxic T-lymphocyte-asscociated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), and programmed cell death protein ligand 1 (PD-L1) also showed limited efficiency in clinical trials,9,10 which may be due to the low tumor mutational burden and nonimmunogenic characteristic of pancreatic tumor.11,12 Thus, it is urgent to explore novel targets for pancreatic cancer therapy.
PDAC tumor microenvironment is mainly composed of fibroblasts, pancreatic stellate cells (PSCs), tumor vasculature, immune cells, and abundant extracellular matrix (ECM). 13 Desmoplastic ECM prevents chemotherapeutic drugs from delivering into the tumor sites 14 and has been shown to be correlated with poor survival. 15 PSCs are one cluster of organ-specific fibroblasts in the pancreas. 16 Activated PSCs acquired myofibroblast-like phenotype 17 and were demonstrated to be capable of upregulating ECM proteins 18 as well as interacting with tumor cells and other cells within tumor stroma to promote tumorigenesis by secreting several factors such as transforming growth factor-β (TGF-β), cytokines interleukin-6 (IL-6) and galectin-119-22 during PDAC progression. Recently, activated PSCs were speculated to be recruited to PDAC epithelium via CC-chemokine ligand 28 (CCL28) secreted by pancreatic tumor cells to remodel the tumor microenvironment, based on the observed CCL28-CCR10 chemotaxis between human PDAC cell line and PSCs in vitro. 23
Upregulated CCL28 was reported to promote tumor progression and metastasis in several types of cancer. Facciabene et al 24 illustrated that tumor hypoxia promotes CCL28 expression and in turn leads to tumor tolerance and angiogenesis via recruitment of regulatory T (Treg) cells in ovarian cancer. Later, CCL28 was also found to play regulator roles in other hypoxic tumors including lung adenocarcinoma and liver cancer.25,26 Of note, hypoxia is a biological feature of pancreatic tumor microenvironment. 27 Studies were also conducted to confirm the anti-apoptosis and metastasis-promoting effects of CCL28 on tumor cells.28,29 However, the effect of CCL28 in PDAC remains poorly defined. The current study elucidated the role of CCL28 in pancreatic cancer and the underlying mechanisms and demonstrated CCL28 as a novel therapeutic target for PDAC.
Materials and Methods
Cell Culture and Generation of
Ccl28
KD Cells
Mouse pancreatic ductal adenocarcinoma cell line PAN02 was purchased from ATCC and cultured in DMEM with 10% FBS and 1% penicillin-streptomycin (Thermo Fisher Scientific). Ccl28-knockdown (Ccl28KD) PAN02 cells were generated with RNA interference. shRNA target sequences against Ccl28 gene (#1: 5′-GCTGTCATCCTTCATGTTAAA-3′; #2: 5′-CCCGCACAATCGTACTTTGAA-3′) were cloned into pLKO.1-TRC cloning vector [Addgene #10878]. PAN02 cells were transduced with lentiviral vectors encoding a nonsilencing scramble shRNA [shScr] sequence or both shCcl28 sequences. After lentiviral infection, PAN02 cells were selected with 2 μg/ml puromycin for 3 days.
Western Blotting
Cell lysates were harvested using RIPA buffer (Beyotime Technology) supplemented with protease inhibitor (Roche) and protein concentration was determined by Pierce BCA Protein Assay Kit (ThermoFisher Scientific). Total proteins (25-30 μg) were applied to 10% to 15% SDS-PAGE and blotted to PVDF membranes (Millipore). Protein bands were visualized using a chemiluminescent HRP substrate (Millipore). Primary antibodies used were anti-CCL28 (Abcam, 1:1000), anti-Bcl-2 (BioLegend, 1:1000), anti-β-catenin (Abcam, 1:5000) and anti-GAPDH (BBI life sciences, 1:1000). Densitometric analysis of blots was performed using ImageJ.
Cell Counting Kit-8 (CCK-8) Assay
Cells were seeded into 96-well plates with a density of 4000 per well and cell proliferation rate was measured by Cell Counting Kit-8 (CCK-8) (Dojindo, Japan) according to the manufacturer's instruction. For cell proliferation rescue, 300 ng/ml recombinant mouse CCL28 (rmCCL28) (BioLegend) was added to the culture medium of PAN02-shCcl28 cells.
5-Ethynyl-2′-Deoxyuridine (EdU) Assay
One million cells were seeded into 6-well plates. When cell confluency was about 80%, cells were incubated with 50 μM EdU (Ribobio) at 37°C for 2 h. Then cultured cells were harvested in single-cell suspension and stained according to the manufacturer's instruction. Fluorescence data were acquired on a BD LSRFortessa cell analyzer (BD Biosciences) and analyzed with FlowJo software.
Transwell Migration and Invasion Assays
A total of 1 × 10 5 cells suspended in serum-free culture medium were seeded in the upper compartment of 8-μm-pore transwell chambers (Corning) with 400 μL complete medium added in the lower compartment. For invasion assay, the 1:20 mixture of Matrigel (Corning) and serum-free medium was plated onto the upper compartment one hour in advance. After 24-h incubation at 37°C, cells passing through the filter were fixed and stained using crystal violet for 10 min. Images of stained cells were collected and the number of migrated or invasive cells were calculated using ImageJ. For each group, 3 and 2 replicated wells were used in migration assay and invasion assay, respectively, 4 fields per well were blindly chosen for counting.
Immunocompetent Syngeneic and Immunocompromised Mouse Tumor Models
Nine-week female C57BL/6J mice supplied by Beijing Vital River Laboratory Animal Technology Co. Ltd were subcutaneously injected with a single-cell suspension of 2 million PAN02 tumor cells suspended in 100 μL phosphate-buffered saline (PBS) per mouse on the right flank as previously described. 30 Tumor length and width were blindly measured with a caliper every 3 days with randomized order.
In the immunodeficient mouse model, eight-week female NOD-SCID mice purchased from Shanghai Shengyin Biotechnology were subcutaneously injected with a single-cell suspension of 2 million PAN02 tumor cells suspended in 100 μL PBS per mouse on the right flank. Tumor length and width were blindly measured with a caliper twice a week with randomized order.
For all animal experiments, tumor volume was calculated as the formula: width2 × length × 0.5. Tumor weights were measured at the endpoints before tumor size reached 500 mm3. Animals were sacrificed by cervical dislocation. Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) and the Bioethics Committee of School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, China (Number: 2020041; Date: September 8, 2020). The reporting of this study conforms to ARRIVE 2.0 guidelines. 31 Animals have been adequately cared for according to the guide. 32 The number of animals utilized was minimized and the suffering of animals was decreased to the minimum under the premise of underpowered studies exclusion. All animals were included without an exclusion criterion.
mRNA Isolation and RT-PCR
Mouse tumor samples were lysed using gentleMACS dissociator (Miltenyi Biotec) according to the manufacturer's instruction. RNA was isolated using the RNeasy Mini Kit (Qiagen). Reverse transcriptase reaction was performed using PrimeScript RT Reagent Kit (Takara). RT-PCR was conducted using SYBR Green PCR Master Mix Kit (Takara). Reactions were run on ABI 7900HT Fast Real-Time PCR System (Applied Biosystems) and relative mRNA expression levels of target genes were calculated by the 2−△△CT methods normalized to internal control Gapdh.
Flow Cytometry
For in vitro analysis, PE-Cy7-conjugated CCR3 antibody (clone J07315, BioLegend) and APC-conjugated CCR10 antibody (clone 248918, R&D Systems) were used to detect cell-surface CCR3 and CCR10 expression. Zombie Green dyes (BioLegend) and BV421-conjugated Annexin V (BioLegend) were used to conduct cell apoptosis assay.
Subcutaneous tumors were dissected and dissociated into single-cell suspension with gentleMACS dissociator (Miltenyi Biotec). The cell suspension was incubated with Zombie Violet dyes (BioLegend) to exclude dead cells before cell-surface marker staining. Nuclear protein Foxp3 was stained using the True-Nuclear Transcription Factor Buffer Set (BioLegend) and APC-conjugated Foxp3 antibody (clone FJK-16s, eBioscience). Antibodies for cell surface markers included BUV395-conjugated CD45 (clone 30-F11, BD Biosciences), PerCP-Cy5.5-conjugated CD3e (clone 145-2C11, BioLegend), BV510-conjugated CD4 (clone GK1.5, BioLegend), PE-Cy7-conjugated CD8a (clone 53-6.7, BioLegend), BV650-conjugated CD335 (clone 29A1.4, BioLegend), PE-conjugated CD11b (clone M1/70, BioLegend), APC-conjugated F4/80 (clone BM8, BioLegend), BV605-conjugated F4/80 (BM8 clone, BioLegend), APC-Fire 750-conjugated Gr1(clone RB6-8C5, BioLegend), PE-Cy7-conjugated Gr1 (clone RB6-8C5, BioLegend), APC-conjugated CD11c (clone N418, BioLegend), PerCP-Cy5.5-conjugated MHC-II (clone M5/114.15.2, BioLegend), and BV510-conjugated CD103 (clone 2E7, BioLegend). Fluorescence data were acquired on a BD LSRFortessa cell analyzer (BD Biosciences) and analyzed with FlowJo software.
Immunofluorescence
Formalin/paraffin-fixed tumor samples were cut into 5-μm sections. Staining was performed using anti-αSMA antibody (Abcam, ab184675, 1:500), anti-CCL28 antibody (Abcam, ab196567, 1:500), and anti-CD31/PECAM1 antibody (Novus Biologicals, NB100-2284, 1:500). Slides were mounted in an anti-fade mounting medium with DAPI (ThermoFisher Scientific) and imaged using a Zeiss fluorescence microscope. Images of 2 random fields per tumor, 4 tumors per group, were chosen for counting. Fluorescence intensity was calculated using ImageJ.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 8.0. For cell proliferation assay, P-value was calculated using two-way ANOVA. In all other assays, the significance of differences between groups was determined by a two-tailed Student's t-test. Significance was defined with P < .05. All data were represented as mean ± SEM.
Results
CCL28 is Upregulated in Pancreatic Cancer and Associated With a Poor Survival Rate
To compare CCL28 expression between PDAC tumors and normal pancreas tissues, we analyzed CCL28 mRNA levels via Gene Expression Profiling Interactive Analysis (GEPIA) public platform 33 and found that CCL28 is significantly overexpressed in PDAC patient samples (Figure 1A) (P < .0001). In addition, CCR3 and CCR10, the known receptors for CCL28, did not show any change between tumors and normal tissues (Figure 1A), suggesting that CCL28 is the major determinant of the activity of this pathway. High CCL28 expression was also found to be significantly associated with poor survival rate in PDAC patients using PROGgeneV2 Pan-Cancer Prognostics Database 34 (Figure 1B). These results implied that CCL28 might contribute to PDAC development and/or progression.
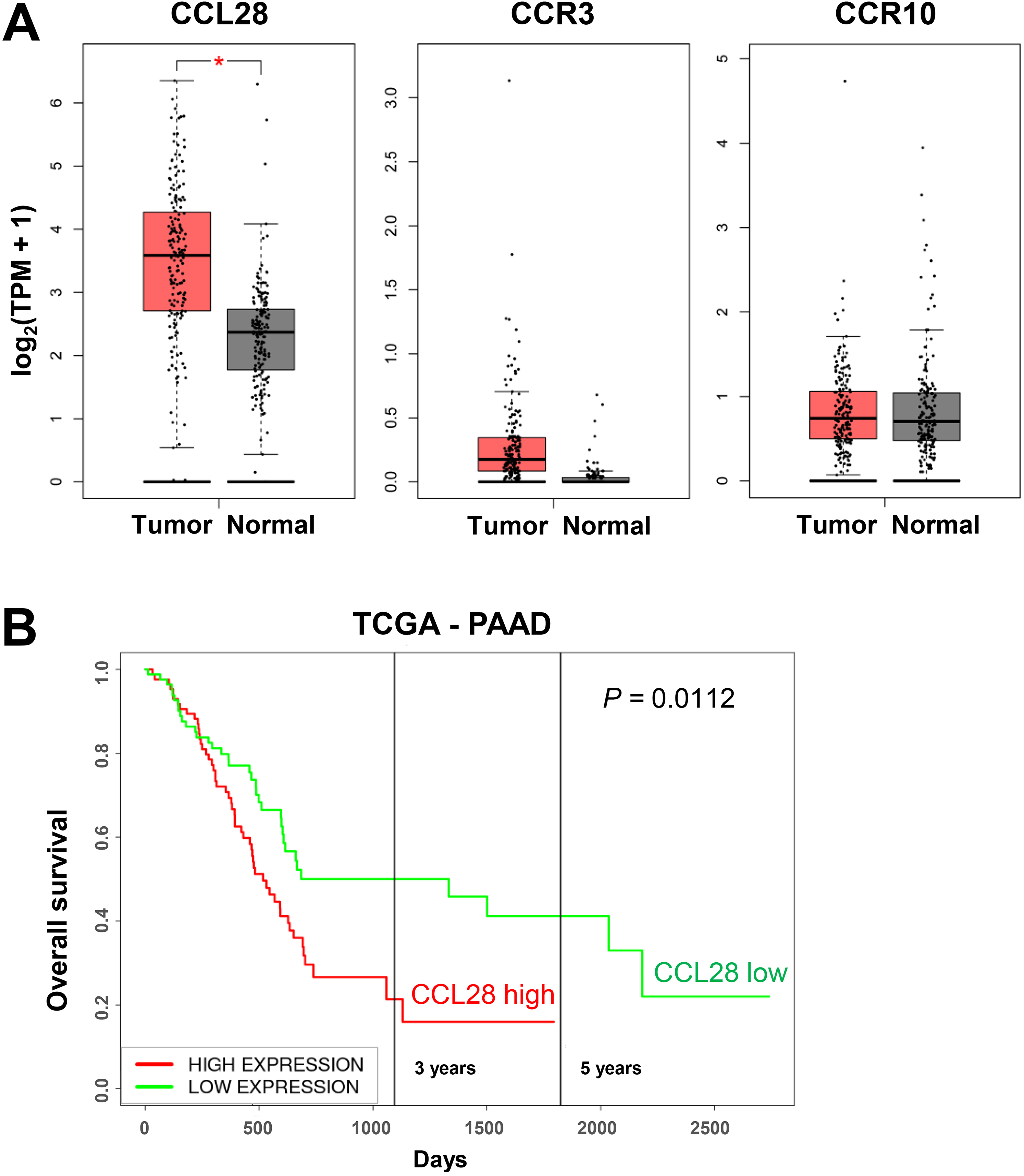
CCL28 is overexpressed in pancreatic cancer and negatively correlated with overall survival. (A) CCL28, CCR3, and CCR10 mRNA expression levels were compared between patient tumor samples (n = 179) and normal pancreas tissues (n = 171) via the Gene Expression Profiling Interactive Analysis (GEPIA) database, *P < .0001; (B) correlation of CCL28 mRNA expression and survival rate in pancreatic cancer patients in TCGA-PAAD (pancreatic adenocarcinoma) dataset.
CCL28 Knockdown Suppresses PDAC Cell Proliferation In Vitro
To explore the cell-autonomous effects of CCL28 in PDAC, we first evaluated the expression of its receptors CCR3 and CCR10 on murine PDAC cell line PAN02. FACS analysis showed that 13% of cells were double positive for CCR3 and CCR10 while only a few cells were single positive (Figure 2A), implying that there might be a co-expression of these 2 receptors. Then CCL28 expression was stably knocked down in PAN02 cells by lentiviral transduction of short hairpin RNA (shRNA) against mouse Ccl28 (shCcl28). Knockdown efficiency was verified by Western blotting (Figure 2B). As determined by the CCK-8 assay, CCL28 knockdown significantly decreased the cell number in comparison to the control group, and this reduction was reversed by the addition of recombinant CCL28 proteins (Figure 2C). To further elucidate whether the change in cell number was due to changed proliferation or apoptosis, we performed an EdU assay and Annexin V staining. A significantly decreased EdU incorporation into Ccl28KD cells indicated a reduced proliferation rate (Figure 2D). In contrast, the ratio of early apoptotic cells was not significantly affected (Figure 2E). Accordingly, the anti-apoptotic factor Bcl-2 was not obviously influenced while a clear downregulation of β-catenin was induced by CCL28 knockdown (Figure 2F). Furthermore, Ccl28KD cells exhibited markedly impaired migratory capability in vitro in the transwell assay (Figure 2G), but no apparent change in cell invasion in the transwell-based invasion assay (Figure 2H). Together, these findings indicated that CCL28-CCR3/10 pathway served as a critical positive regulator of PDAC cell proliferation in vitro.

CCL28 knockdown impairs pancreatic cancer cell proliferation and migration in vitro. (A) Cell surface expression of CCR3 and CCR10 was detected by flow cytometry in PAN02 cells; (B) immunoblotting image and quantification of CCL28 in PAN02 cell transduced with lentiviral shRNA (n = 3); (C) cell proliferation was measured in shCcl28 (Ccl28KD) cells with or without exogenous mouse recombinant CCL28 and shScr control cells using the CCK-8 assay (n = 4); (D and E) EdU-based cell proliferation assay (D) and cell apoptosis assay (E) were performed in shCcl28 (Ccl28KD) and shScr control cells using flow cytometry (n = 3); (F) immunoblotting images and quantification of Bcl-2 and β-catenin in shCcl28 (Ccl28KD) and shScr control cells (n = 3); (G) cell migration was evaluated by transwell assay (n = 12); (H) cell invasion ability of shCcl28 cells and shScr control cells was determined (n = 8). Scale bar = 100μm; * P < .05, ** P < .01, *** P < .001, **** P < .0001, n.s. = not significant.
CCL28 Downregulation Promotes Tumor Control in Mouse Models
We then sought to study the effects of CCL28 on PDAC tumor growth in vivo. First, we established subcutaneous tumors using Ccl28KD (shCcl28) or control (shScr) PAN02 cells in immunodeficient NOD-SCID mice with the absence of T and B lymphocytes and compromised functions of NK cells, macrophages, and dendritic cells (DCs) to exclude the effects of immunosurveillance (Figure 3A). Clearly, CCL28 deficiency led to reduced tumor growth, probably via the effects on tumor cell proliferation. Consistently, the tumor weights at the endpoint were significantly lower in the shCcl28 group (Figure 3A). Furthermore, we established mouse PDAC syngeneic models by subcutaneous injection of shCcl28 or shScr PAN02 cells. According to the tumor growth curve, CCL28 knockdown significantly repressed tumor progression since day 9 after tumor inoculation and the anti-tumor effect became more prominent until the last day of monitoring (Figure 3B). These data demonstrated an evident tumor-inhibiting role of CCL28 downregulation.

CCL28 knockdown inhibits pancreatic tumor growth. In NOD-SCID mice (A) and C57BL/6J mice (B), volumes of shCcl28 (Ccl28KD) and shScr subcutaneous tumors were monitored overtime (n = 9 - 10/group) and tumor weights were measured at the endpoint time. Scale bar = 1 cm; * P < .05, ** P < .01, *** P < .001.
CCL28 downregulation shapes tumor immune microenvironment
Since CCL28 has been shown to regulate other cell types besides tumor cells in the tumor microenvironment,23,24 mechanisms other than its effect on tumor cell proliferation may be also involved in pancreatic cancer progression. To gain more mechanistic insight into how CCL28 deficiency inhibits PDAC tumor progression, we next examined the intratumoral infiltration of immune cells by flow cytometry in an immunocompetent syngeneic mouse model. Inconsistent with previous reports in other cancer types,24,25,35 CCL28 downregulation notably suppressed Foxp3+ Treg cells accumulation (Figure 4A). Ratios of infiltrated Foxp3− conventional CD4+ T cells and cytotoxic CD8+ T cells showed no differences between the 2 groups (Figure 4A and B). Moreover, the ratio of NK cells showed a nearly significant elevation in Ccl28KD tumors (Figure 4C). Besides Treg cells, we also detected another immunosuppressive cell population, the myeloid-derived suppressor cells (MDSCs), and observed an evident decrease in the ratio of MDSCs in CCL28-deficient tumors (Figure 4D). Interestingly, a reciprocal regulation between MDSCs and Treg cells has been reported in PDAC. 36 In contrast, no change in the ratios of other myeloid cells including macrophages, CD103+ or CD11b+ conventional DCs (cDCs) was observed (Figure 4E and F).

CCL28 downregulation remodels pancreatic tumor immune microenvironment. FACS was conducted to detect tumor-infiltrating immune cells in shCcl28 (Ccl28KD) and shScr control PAN02 tumors. Percentages of Treg cells and conventional Foxp3− CD4+ T cells (Conv. CD4+ T) (A), CD8+ T cells (B), NK cells (C), MDSCs (D), macrophages (E), and cDCs (F) in CD45+ cells were shown. * P < .05, *** P < .001.
In addition, we measured the expression of different cytokines and factors in the tumors. qPCR results revealed an increase in the expression of anti-tumor cytotoxic proteins including granzyme B (Gzmb) and perforin (Prf1), but no change in the expression of anti-tumor cytokine IFN-γ (Ifng), or a variety of interleukins including the immunosuppressive cytokine IL-10 (Il10) (Figure 5). The angiogenic factor VEGF was decreased in the Ccl28KD tumors (Figure 5).

Quantification of cytokine expression in PAN02 tumors in syngeneic mouse model. mRNA expression levels of cytokines including granzyme B (Gzmb), perforin (Pfr1), IFN-γ (Ifng), TNF-α (Tnf), vascular endothelial growth factor A (Vegfa), and the interleukin (Il) family members, were measured by RT-PCR in tumor samples (n = 7-9). * P < .05, **** P < .0001.
In brief, CCL28 downregulation could relieve the immunosuppressive state by decreasing the infiltrations of Treg cells and MDSCs, and enhance anti-tumor immunity by upregulating cytotoxic proteins that were mainly derived from CD8+ T and NK cells.
CCL28 Deficiency Leads to Decreased Infiltration of Pancreatic Stellate Cells and Blood Vessel Formation
Previous studies have proposed a role of CCL28 in mediating the migration of activated PSCs in PDAC, 23 and a critical pro-tumoral role of PSCs via inhibiting lymphocyte function and recruiting MDSCs.19,20,37 Finally, we examined the frequency of activated PSCs in subcutaneous tumors through immunostaining of its activation marker alpha-smooth muscle actin (α-SMA). 38 Immunofluorescent results confirmed a downregulation of CCL28 in the tumor (Figure 6A) and revealed a significantly lower level of α-SMA in the Ccl28KD tumors (Figure 6B), indicating a less enrichment of activated PSCs. Therefore, targeting CCL28 indeed impeded the migration of activated PSCs into PDAC tumors. In agreement with decreased VEGF expression (Figure 5), Ccl28KD tumors exhibited less CD31 staining (Figure 6C), implying that CCL28 knockdown resulted in reduced blood vessel formation.
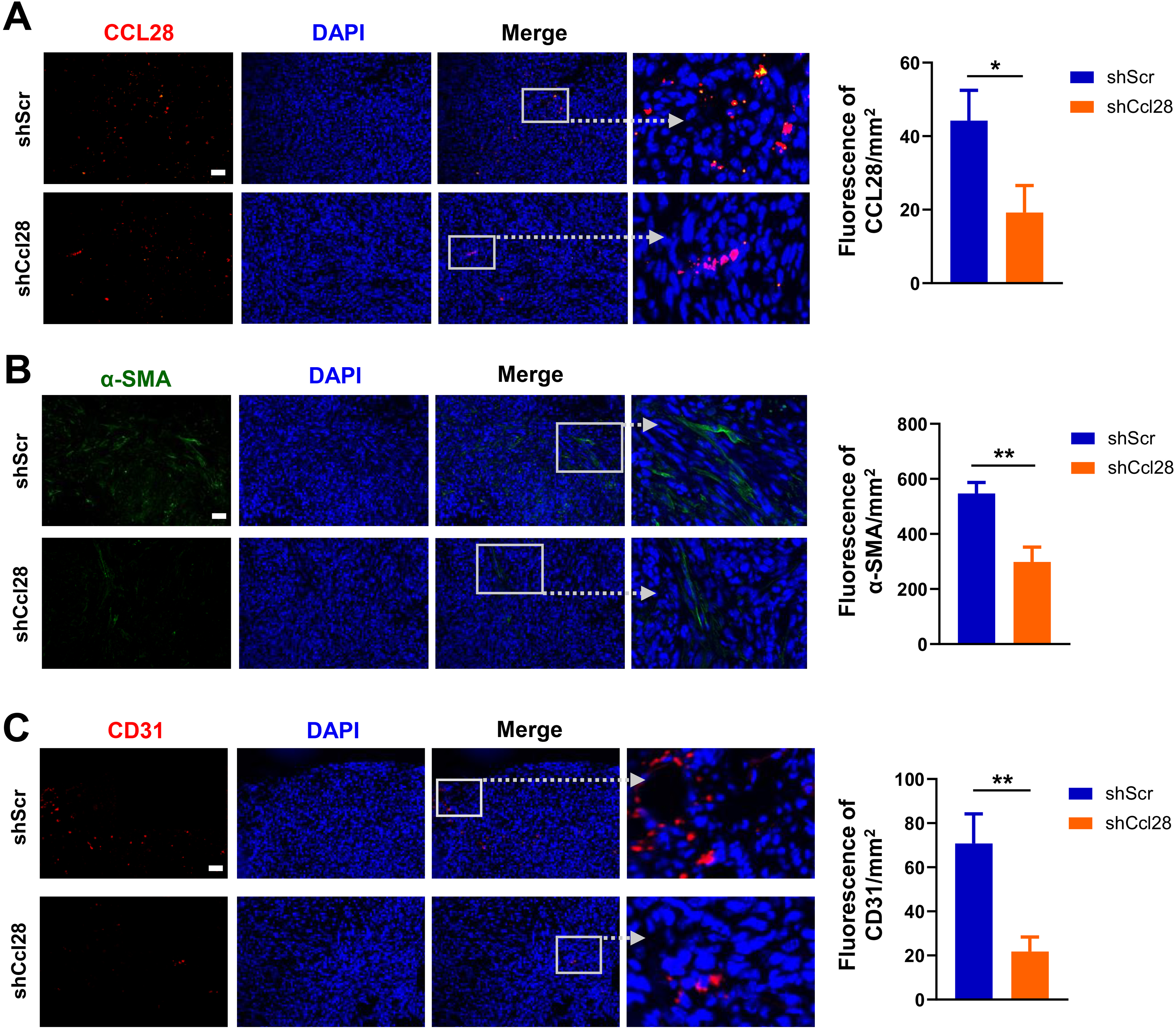
CCL28 downregulation inhibits tumor infiltration of activated PSCs and angiogenesis. Representative images of immunofluorescence (IF) and quantification for CCL28 (A), α-SMA (B), and CD31 (C) were shown (n = 8). Scale bar = 100 μm; * P < .05, ** P < .01.
Discussion
In the current study, we elucidated the complex role of CCL28 in regulating pancreatic cancer progression (Figure 7). CCL28 primarily functions as a chemokine to control cell migration. Accordingly, our results have shown that CCL28 regulates the migration of pancreatic tumor cells, Treg cells, and PSCs. Moreover, it also stimulates pancreatic tumor cell proliferation, in consistence with the observation that CCL28 enhances breast cancer cell proliferation. 28 Therefore, CCL28 can promote pancreatic cancer progression through an autocrine loop and the tumor cell-intrinsic mechanisms, by influencing tumor cell proliferation and migration. However, it remains to be investigated whether CCR3 and CCR10 differentially regulate the tumor cells in response to CCL28 stimulation and whether their expression marks the cell population with higher proliferative and/or migratory capacity.

Schematic model illustrating the mechanisms for CCL28 inhibition-induced PDAC tumor suppression. On the one hand, CCL28 controls tumor cell proliferation in an autocrine way. On the other hand, CCL28 inhibition shapes tumor microenvironment via decreasing the accumulation of Treg cells, PSCs, and MDSCs while upregulating the expression of anti-tumor cytotoxic proteins including perforin and granzyme B.
In addition to tumor cell-intrinsic mechanisms, the tumor cell-extrinsic microenvironment also plays a critical role in cancer development and progression. Immunosuppressive cells such as Treg cells, MDSCs, and cancer-associated fibroblasts (CAFs) have strong pro-tumoral effects, thus serving as potential therapeutic targets. In pancreatic tumors, CAFs mainly derive from PSCs which differentiate into myofibroblast-like cells upon activation and play multifaceted roles.17,39 Our data demonstrated that downregulation of CCL28 reduced the infiltration of these cells in pancreatic tumors. Consequently, anti-tumor activity was enhanced by CCL28 knockdown as indicated by the upregulation of lymphocyte-restricted cytotoxic proteins including perforin and granzyme B in the tumors. Moreover, impaired angiogenesis was illustrated by lower VEGFA and CD31 levels in CCL28-deficient tumors, which may be related to decreased MDSCs infiltration, considering their pro-angiogenesis role in PDAC. 40 The direct control of migration of Treg cells and PSCs by CCL28 has been verified,23,24,35 although it is still unknown whether CCL28 regulates MDSC migration directly. It's possible that the observed decrease of MDSCs in Ccl28KD tumors depended on the effect of PSCs since PSCs have been shown to promote MDSC differentiation. 19 Alternatively, the mutual interaction between Treg cells and MDSCs may be also involved.36,41,42 Furthermore, the contribution of each immunosuppressive cell type to CCL28-induced tumor progression remains to be clarified.
Conclusion
Given the fact that CCL28 is negatively associated with the survival of pancreatic cancer patients and promotes tumor growth via various mechanisms, it is potentially a very promising therapeutic target. Blockade or downregulation of CCL28 is able to elicit both tumor cell-intrinsic and -extrinsic anti-tumor activities in pancreatic cancer, for example, inhibition of tumor cell proliferation and accumulation of immunosuppressive cells including Treg cells, MDSCs, and PSCs, respectively. Targeting CCL28 may also synergize with many other immunotherapies since these immunosuppressive cells quite often limit the efficacies of immunotherapies.
Data Availability
All data included in this study are available upon request by contact with the corresponding author.
Footnotes
Authors’ Contribution
JY and BM designed the experiments; JY, LG, PYuan, ZW, and PYin conducted the research; JY and BM analyzed the data; JY, W-QG, and BM wrote the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by funds from Science and Technology Commission of Shanghai Municipality (20ZR1426500 to BM, 20JC1417600 to W-QG), and the 111 Project (B21024).
Study Approval
All animal procedures were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University, Shanghai, China (Number: 2020041; Date: September 8, 2020).