
Abstract
Introduction
All cells in the human body contain the same DNA blueprint, but alterations in the regulation of gene expression are frequently associated with development, differentiation, as well as the pathogenesis of diseases. The expression of genetic information of a cell starts with transcription, which is followed by posttranscriptional modification. Dysregulation of both processes is the hallmark of cancer.1–3 Posttranscriptional regulation, which includes pre-mRNA splicing and polyadenylation, mRNA transport, and translation, mostly occurs through the action of RNA binding proteins (RBPs) and processing factors. These molecules associate with RNAs from the initiation of transcription to the eventual death of RNAs in the cytoplasm.4–6 As a key player in posttranscriptional events, RBPs modulate multiple cancer traits by forming an interactive network with RNA. 7 Through RBPs, cancer cells may sustain their proliferation by altering pathways that regulate alternative splicing. 8 The high frequencies of mutations or altered expression and/or the activity of splicing factor proteins and spliceosome components have been identified in multiple tumor types and disease states. Notably, many SR proteins, hnRNPs, and other splicing factors play protumorigenic or antitumorigenic roles. 9 Many splicing factors exhibit dysregulated expression in cancer. These are associated with altered splicing patterns in diseased tissues, 10 tumor stages, 11 metastatic potential, or poor prognosis.12,13 In addition, recurrent spliceosomal mutations usually alter splice sites or exon recognition preferences to cause abnormal, cancer-specific RNA splicing. 9 High frequencies with which SF3B1, U2AF1, and SRSF2 are subject to specific recurrent mutations in hematological malignancies suggest that spliceosomal mutations can drive tumorigenesis.14–16 Recent progress in our understanding of transcription and posttranscriptional regulation and its role in cancer pathogenesis suggests that many new insights will soon be leveraged for the benefit of the patient.
The improvement in mRNA interactome capture technology allows researchers to more comprehensively identify RBPs that play key roles in cancer pathogenesis. At present, more than 1000 RBPs have been identified based on high-throughput omics technologies. 17 These RBPs affect all RNA metabolic processes, including splicing, transport, translation, and decay. ATP5A1, for example, is a recently identified RBP generally regarded to mediate tumor progression through mitochondrial signaling. In 2012, Castello et al 18 first identified the RNA binding function of ATP5A1 by applying a systematic and unbiased mRNA interactome capture approach using human HeLa cells. However, the functions of ATP5A1 as a novel RBP are rarely studied and largely unknown.
ATP5A1 (also named ATP5F1A), which encodes for a subunit of ATP synthase (complex V), is overrepresented in an oxidative phosphorylation pathway that is associated with tumorigenesis and tumor progression. The mitochondrial ATP synthase is composed of the soluble catalytic core, the membrane-spanning component, F0, and ATP5A1 as the alpha subunit of F1.19,20 ATP is produced from ADP by ATP synthase in the presence of a proton gradient across the membrane. This gradient is generated by electron transport complexes of the respiratory chain. 21 Mitochondrial ATP production is the main energy source for intracellular metabolic pathways. 22 Defects or mutations affecting the subunits in ATPase synthase are known to cause many diseases in humans. 23 Mutations in the α subunit, for example, have been associated with neuropathy, ataxia, retinitis pigmentosa syndrome, familial bilateral striatal necrosis, and one form of the Leigh syndrome.24,25 Tumors arise, progress, and respond to therapy in the context of intimate crosstalk with the host immune system. Similarly, many immunological functions rely on reprogrammed mitochondrial energy metabolism. This is achieved by boosting aerobic glycolysis as the main pathway for the higher metabolic energy requirements and production of precursor molecules for biosynthetic purposes.26,27 Accordingly, the expression of the catalytic subunit of the ATP synthase is dysregulated in human carcinomas. 28 Downregulation of many ATP synthase subunits, including ATP5A1, was observed in clear cell renal cell carcinoma (ccRCC) cases, thereby suggesting its prognostic value in ccRCC patients.29,30 It was demonstrated that ATP5A1 is overrepresented in oxidative phosphorylation pathways that are associated with tumorigenesis and tumor progression. The dysregulated expression of ATP synthase subunits could be the basis for the reduced activity of the mitochondrial electron chain and reduced oxidative phosphorylation. Most probably caused by the downregulation of miRNAs that target ATP5A1 and ATP5B, glioblastoma (GBM) tumor cells have similarly been found to have significantly higher expression of ATP5A1 and ATP5B, as well as microvascular proliferation. 31 Therefore, miRNAs that target ATP5A1 or ATP5B might be potential therapeutic agents for GBM.
Seth et al 32 proposed that higher levels of ATP5A1 were associated with certain SNPs and with TP53 mutations. Moreover, high expression of ATP5A1 occurs in chromosomal instability and may facilitate tumor development along this pathway. Conversely, low levels of ATP5A1 may facilitate the development of tumors with microsatellite instability. 32 A previous study showed that ATP5A1 is a biomarker of inner ear exosomes, 33 suggesting its potential involvement in ear function and diseases, including ear cancer. Malignancy of the ear is a rare entity among all head and neck tumors, with an estimated incidence of 1 case per million people in the world.34,35 According to the histologic features, ear tumors mainly include squamous cell carcinoma (SCC), ceruminous neoplasms, papillary tumors, endolymphatic sac tumors, otosclerosis, and cholesteatoma, which all occur in the external auditory canal or the middle or inner ear. 34
To systematically explore the transcriptional and posttranscriptional regulation of ATP5A1, we enhanced its expression in HeLa cells by transfecting a plasmid with ATP5A1 cDNA sequences. Whole transcriptome sequencing analysis using RNA-seq was then performed to identify ATP5A1-regulated gene expression changes and alternative splicing events (ASEs). The results suggest that ATP5A1 serves potentially important roles in regulating gene expression and alternative splicing of cancer-related pathways. This extends our understanding of the function of ATP5A1 in cancer.
Materials and Methods
Cell Culture and Transfections
Deriving from a human cervical cancer cell line, HeLa cells (CCTCC@GDC0009) were obtained from the CCTCC (China Center for Type Culture Collection) in 2017. The obtained cells were authenticated with STR analysis by the Cell Bank, Type Culture Collection, Chinese Academy of Sciences (CBTCCCAS) and tested to ensure that the samples were free of mycoplasma contamination upon receipt from the provider. Genomic DNA was isolated with Purelink@ Genomic DNA Kits in the Cell Bank, and then analyzed at the Beijing Microread Genetics Co., Ltd. The samples were amplified using the Goldeneye™ 20A STR Complex Amplification Kit. The profiles of the STR loci and Amelogenin gene were characterized on an ABI 3100 Genetic Analysis Instrument.
HeLa cells were cultured with 5% CO2 at 37 °C in DMEM (Dulbecco's modified Eagle's medium), with 10% FBS (fetal bovine serum), 100 U/mL penicillin, and 100 µg/mL streptomycin. To overexpress ATP5A1 in HeLa cells, an ATP5A1 cDNA-containing plasmid was constructed using the pGFP-B-RS vector. Primer pairs used for hot fusion reactions were designed with a 17 to 30 bp overlap between primer pairs using CE Design v1.04 (Vazyme Biotech Co., Ltd). Gene-specific sequences were targeted for the primer design and also included a portion of the pIRES-hrGFP-1a vector sequences. The primer sequences were as follows: forward, 5′-AGCCCGGGCGGATCCGAATTCATGCCGCGCGTCTACATAGG-3′ and reverse, 5′-GTCATCCTTGTAGTCCTCGAGATCTCTGGAACTCGACCTGGACC-3′. The ATP5A1 overexpression plasmid was constructed and transfected into HeLa cells. Using a previously published method, empty control plasmids were also transfected. 36
Assessment of the Overexpression of
ATP5A1
The GAPDH (glyceraldehyde-3-phosphate dehydrogenase) housekeeping gene was used as an internal control gene to assess the effects of ATP5A1 overexpression. cDNA synthesis was performed by standard procedures following real-time quantification PCR. The latter was performed using the HieffTM qPCR SYBR® Green Master Mix (Low Rox Plus, YEASEN) to evaluate the expression level of ATP5A1. The information relating to the primers used for the RT-qPCR is presented in Supplemental Material. The expression of transcripts was then compared against GAPDH mRNA levels using the 2−ΔΔCT method. 37 Using a previously described western blot (WB) assay, 38 the ATP5A1 protein level was also assessed. The following primary antibodies were used: antiflag (1:1000 dilution; polyclonal antibody; cat. no. 2368S; CST) and antiactin (1:1000 dilution; polyclonal antibody; cat. no. AC026; ABClonal). The following secondary antibody was used: horseradish peroxidase-conjugated goat antirabbit IgG (1:1000 dilution; cat. no. AS014; ABClonal). The slightly cropped image of the WB result was provided in Figure S1.
Cell Proliferation and Apoptosis Assay
The MTT assay was used to measure cell proliferation. Prior to the experiment, HeLa cells (1 × 104) were seeded into 96-well culture plates with 200 μL of the cell growth medium. Cellular apoptosis levels were assessed using flow cytometry. Detailed experimental procedures describing the cell proliferation and apoptosis assays have been presented in a previous study. 38
RNA Extraction and High-Throughput Sequencing
Total RNA was extracted using the TRIZOL (Ambion) method and was further purified with 2 phenol-chloroform treatments. Genomic DNA was removed using RQ1 DNase (RNase free; Promega) to obtain RNA. Quality and quantity of purified RNA were obtained by checking the absorbance at 260/280 nm (A260/A280) using a Smartspec Plus (BioRad) spectrophotometer. The integrity of RNA was then verified by 1.5% agarose gel electrophoresis.
For each sample, we used 10 μg total RNA to prepare a unidirectional RNA-seq library. Oligo(dT)-conjugated magnetic beads (Invitrogen) were used to enrich polyadenylated mRNAs prior to library prep. RNAs used for library construction were prepared according to a previously published study. 39 The libraries were prepared according to the manufacturer's instructions for high-throughput sequencing. The Illumina HiSeq4000 system was used to collect data from 151-bp pair-end sequences (ABlife Inc.).
Cleaning and Alignment of Raw RNA-seq Data
Raw sequencing reads with more than 2 N bases were discarded first. Adaptors and low-quality bases were then trimmed using the FASTX-Toolkit (Version 0.0.13), whereafter short reads (reads <16 nt) were finally removed. The quality-filtered reads were subsequently aligned to the GRCH38 genome using TopHat2. 40 No more than 4 mismatches were permitted per read. Uniquely mapped reads were ultimately used to calculate the read number and FPKM (paired-end fragments per kilobase of exon per million fragments mapped) value for each gene.
Differentially Expressed Gene Analysis
EdgeR 41 was used to identify the differentially expressed genes (DEGs) using RNA-seq data. Fold change (fold change ≥2 or ≤0.5) and P-value (P-value < 0.01) criteria were used to determine whether a gene was differentially expressed.
Gene function and the frequency distribution of each category were predicted using KOBAS 2.0 server 42 and Gene Ontology (GO) and KEGG pathway enrichment analyses. The enrichment of each pathway (corrected P-value < 0.05) was defined using the hypergeometric test and Benjamini–Hochberg FDR procedure.
Alternative Splicing Analysis
The ABLas pipeline, as described previously, 43 was used to define and quantifyASEs and regulated ASEs (RASEs) across the samples. In brief, the detection of 7 types of canonical ASEs in each sample was based on the splice junction reads. These ASEs included exon skipping (ES), cassette exons (CE), alternative 5′ splice sites (A5SS), alternative 3′ splice site (A3SS), mutual exclusive exon skipping (MXE), MXE combined with an alternative polyadenylation site (3pMXE), and MXE with an alternative 5′ promoter (5pMXE).
P-values were subsequently calculated using the Fisher exact test, where the modeled sample reads and alternative reads served as input data, respectively. Defined as the RASE ratio, the change in the ratio between alternatively spliced reads and constitutively spliced reads was calculated across samples. A RASE ratio >0.2 and P-value < .05 were set as the threshold for RASE detection.
Quantitative Reverse-Transcription PCR Validation of ASEs
To elucidate the validity of DEGs and ASEs in HeLa cells, quantitative reverse-transcription polymerase chain reactions (RT-qPCRs) were performed for the selected DEGs and RASEs and normalized against the GAPDH reference gene. The primers used for detecting the pre-mRNA splicing sequences are shown in Table S3. The detailed method is fully described in a previously published study. 44
Downloading Cervical Cancer RNA-seq Data
The expression of ATP5A1 was analyzed in datasets from multiple cancers obtained from the TCGA (the cancer genome atlas) database. The detailed expression patterns and survival time analysis of ATP5A1 in cervical squamous cell carcinoma (CESC) were analyzed using GEPIA2. 45
Results
The Expression Profile of the ATP5A1 Gene Showed Variable Patterns in Cancers
The expression profile of ATP5A1 was explored in multiple cancers by extracting the TCGA transcriptome profile. Among the 31 cancer types examined, as shown in Figure 1a, ATP5A1 showed nonuniform expression patterns with 6 significantly increased cancer types (CHOL, KICH, LIHC, LUSC, SKCM, and UCEC) and 10 significantly decreased cancer types (BLCA, BRCA, COAD, ESCA, HNSC, KIRC, KIRP, READ, STAD, and THCA). This suggests that ATP5A1 has multiple functions in the aforementioned cancers. Inspired by the ability of ATP5A1 to associate with RNAs, 18 the capacity of ATP5A1 to regulate the RNA expression and alternative splicing in cancer cells beyond its ATP synthesis function was examined. HeLa cells were chosen as the target cell line because of their wide application in cancer research. Before performing the experiments, the expression and prognostic signature of ATP5A1 were examined in the cervical squamous cell carcinoma (CESC) data. The expression pattern of ATP5A1 in these cells showed a slightly higher level in stage1 to 4 patients during CESC progression (Figure 1b). However, CESC patients with higher ATP5A1 expression showed better prognostic signatures (Figure 1c), thereby demonstrating the tumor suppression function of ATP5A1 in CESC.
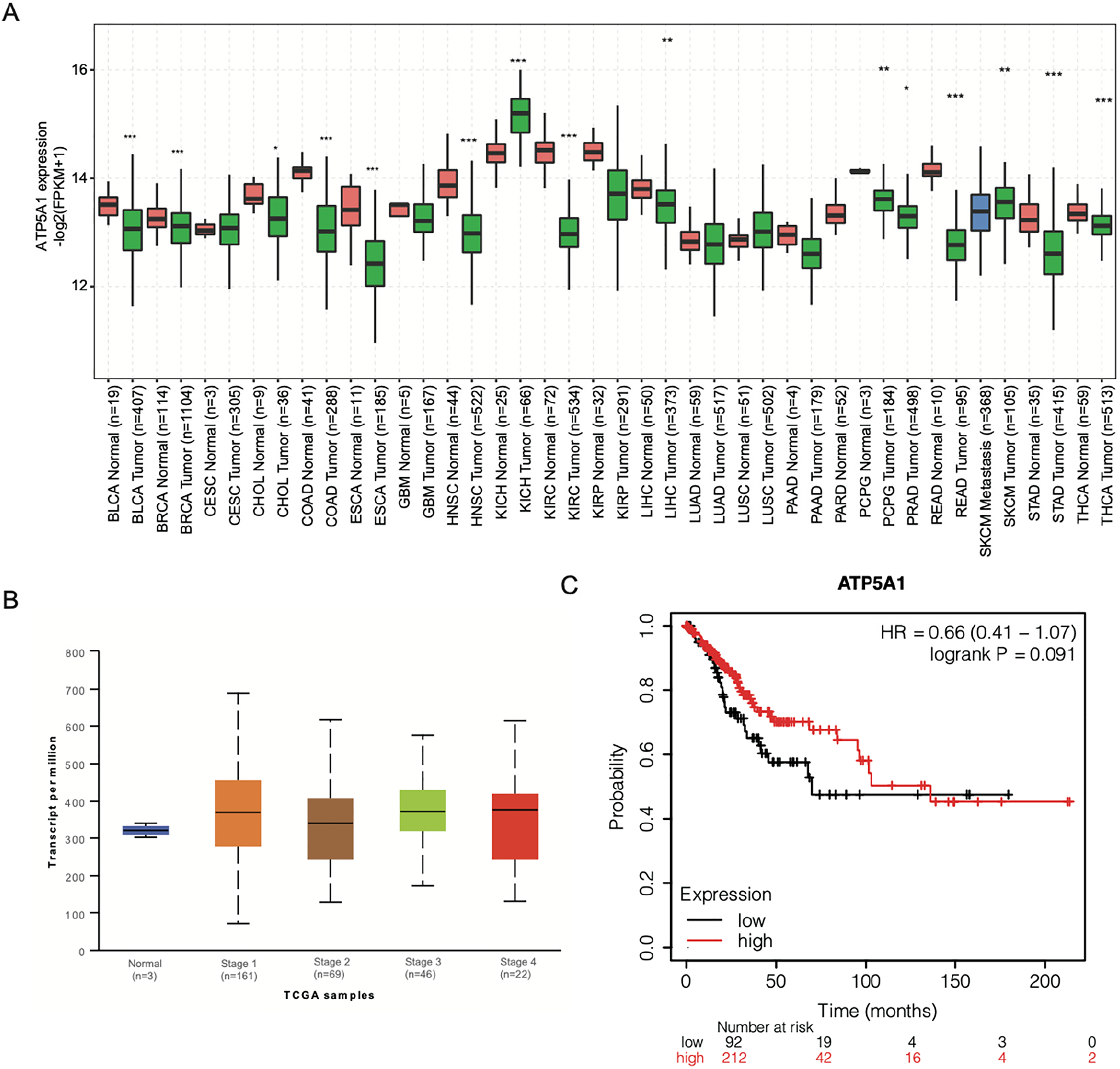
Expression pattern of ATP5A1 in TCGA data and cervical squamous cell carcinoma (CESC) patients. (a) Box plot showing the expression pattern of ATP5A1 in multiple cancer types from data obtained from the TCGA transcriptome database. Expression differences were assessed using Student's t-test (* P < 0.05; ** P < 0.01; *** P < 0.001). (b) Box plot showing the detailed expression level between healthy samples and cervical tissue samples derived from CESC patient tumors across the 4 stages. (c) Line graph showing the survival time of CESC patients divided into 2 groups according to ATP5A1 expression level.
ATP5A1 Participates in Transcriptional Regulation in HeLa Cells
Based on the initial analysis of the TCGA data, ATP5A1 expression was increased in HeLa cells by transfecting a plasmid with ATP5A1 sequences into HeLa cells, while simultaneously preparing a negative control. RT-qPCR and WB analysis both validated the success of the overexpression experiment (Figure 2a and b). After overexpression, it was found that ATP5A1 could significantly increase the level of apoptosis in HeLa cells (Figure 2c), but it had no influence on cell proliferation (Figure S2A). These results revealed that ATP5A1 could induce cellular apoptosis processes in HeLa cells. To uncover the underlying mechanism of ATP5A1 on cellular changes, whole transcriptome analysis (RNA-seq) was performed on ATP5A1 overexpression (OE) and negative control (NC) cells. 2 biological replicates were prepared for ATP5A1-OE and NC cells, with about 84.3 million quality-filtered reads being obtained per sample (Table S1). The reads were then aligned to the GRCH38 genome using TopHat2, 40 and the expression level for each gene identified by calculating the FPKM value. The FPKM value of ATP5A1 also confirmed its successful overexpression in HeLa cells (Figure 2d). The Pearson correlation was used to assess the global similarity between ATP5A1-OE and NC samples. With the exception of the overexpressed ATP5A1 gene, the analysis demonstrated that ATP5A1-OE and NC samples showed a high level of similarity for all expressed genes (R = 0.993; Figure 2e), implying that ATP5A1 overexpression changed the transcriptome profile in a limited scope of HeLa cells. Using differentially expressed gene (DEG) analysis, genes significantly regulated by ATP5A1 were then explored. With a 2-fold change in expression and 0.01 P-value as the filtering criteria, 590 DEGs were found between ATP5A1-OE versus NC samples. This included 309 downregulated and 281 upregulated DEGs (Figure 2f). A hierarchical clustering heatmap of the expression pattern of the 590 DEGs showed that they have a consistent expression pattern in ATP5A1-OE and NC cells (Figure 2g). Excluding the upregulation of ATP5A1, HSPA6, HSPA1A, FTH1P10, HSPA1B, and FBXW10 were found to be upregulated and with the most significant P-values (Table S2). 3 of these DEGs (HSPA6, HSPA1A, and HSPA1B) belong to the heat shock protein 70 family. To investigate how ATP5A1-OE promoted apoptosis, expression levels of genes in the mitochondrial apoptosis pathway were analyzed. 46 Although proapoptotic genes were increased and, with the exception of BCL2L1, antiapoptotic genes were decreased in ATP5A1-OE samples, these DEGS were not significantly different (Figure 2h). This implied that ATP5A1 has an underlying proapoptosis function.

RNA-seq analysis revealed the global transcriptome change by ATP5A1-OE. (a) Western blot showing the successful overexpression of ATP5A1 in HeLa cells. (b) Bar plot showing the significant overexpression of ATP5A1 by RT-qPCR (Student's t-test, ** P < 0.01). (c) Flow cytometry dot plots (upper panel) and bar plot (bottom panel) showing the increased apoptosis level in ATP5A1-OE cells. 3 biological replicates were tested to obtain the result. (d) Bar plot showing the significant overexpression of ATP5A1 by RNA-seq values (Student's t-test, ** P < 0.01). (e) Dot plot showing the high correlation between ATP5A1-OE and NC samples. The outlier, ATP5A1, is indicated by the red dot. (f) Volcano plot showing the differentially expressed genes between ATP5A1-OE and NC samples. Red and blue points represented upregulated and downregulated genes, respectively. (g) Hierarchical clustering heat map showing the expression pattern of DEGs between ATP5A1-OE and NC samples. (h) Hierarchical clustering heat map showing the expression level of genes involved in the mitochondrial apoptosis pathway between ATP5A1-OE and NC samples.
ATP5A1 Positively Regulates the Expression of Genes Enriched in Angiogenesis and Immune Response
To explore the biological influences of ATP5A1 overexpression in HeLa cells, functional enrichment analysis was performed separately for upregulated and downregulated DEGs. Focusing on the upregulated genes associated with ATP5A1-OE samples, the collagen catabolic process, angiogenesis, and small molecule metabolic processes were found to be the most enriched GO biological pathway (BP) terms. 2 genes, MMP2 and MMP19, were involved in both the collagen catabolic process and angiogenesis terms. Compared to MMP19, expression levels of MMP2 were much lower in both ATP5A1-OE and control samples (FPKM <0.1). The innate immune response was also within the top ten enriched GO BP terms (Figure 3a). The estrogen signaling pathway was noted as the most enriched pathway following KEGG analysis (Figure 3b). For downregulated genes, enrichment was observed in the regulation of transcription (DNA-dependent), inflammatory response, and cell adhesion BP terms following GO analysis (Figure S2B). KEGG analysis reported that fatty acid biosynthesis, taste transduction, and cytokine–cytokine receptor interaction pathways were the most enriched (Figure S2C). To validate the DEG analysis results, 8 DEGs were selected for RT-qPCR experiments. This included 6 upregulated (CLDN9, FCER1G, HSPA1A, HSPA1B, HSPA6, and MMP19) and 2 downregulated (CCL26 and GH1) DEGs. The RT-qPCR results showed that the selected DEGs were also significantly regulated by ATP5A1, which was highly consistent with the RNA-seq results (Figure 3c and Figure S2D). These results demonstrated that ATP5A1 could regulate the expression of genes in multiple cancer-related pathways.

DEG functional analysis and validation experiment. (a) Bubble plot showing the top ten enriched GO BP terms for ATP5A1-OE-associated upregulated DEGs. Terms related to cancer pathways are labeled in red and blue. (b) Bubble plot showing the top ten enriched KEGG pathways for ATP5A1-OE-associated upregulated DEGs. (c) Bar plot showing the RNA-seq and RT-qPCR validation results for 6 selected DEGs.
ATP5A1 Selectively Regulates the Alternative Splicing of Cancer-Related Genes
Based on the fact that aberrant alternative splicing can promote growth and survival of cancer, 47 we wanted to see whether the tumor-related functions of ATP5A1 were associated with alternative splicing regulation. By using TopHat2 software annotation, 40.07% to 43.26% of the aligned reads presented with evidence of splicing (Table S1). ATP5A1-OE samples had a higher ratio of spliced reads (Figure 4a). To further explore the ATP5A1-regulated alternative splicing events (RASEs), the ABLas method was used to systematically identify RASEs between ATP5A1-OE and NC cells. With a P-value < 0.05 as the threshold, 641 RASEs were identified. This included 295 upregulated and 346 downregulated ASEs (Figure 4b). The most prominent forms of ASEs observed included A5SS, A3SS, ES, and CE (Figure 4b). Notably, CE events were specifically repressed in ATP5A1-OE samples (Figure 4b). To assess the relationship between gene expression and alternative splicing, the DEGs were overlapped with regulated alternative splicing genes (RASGs). As only 1 overlapping gene was found (Figure 4c), ATP5A1-regulated gene expression and alternative splicing were identified as representing 2 independent processes. Functional enrichment analysis was then performed for the RASGs. GO BP enrichment analysis revealed that glucose homeostasis and response to calcium ions were the top enrichment terms. This was followed by 3 terms involving transcriptional regulation and the cellular response to hypoxia (Figure 4d). KEGG pathway enrichment analysis revealed that the 1 carbon pool by folate, mTOR, and HIF-1 signaling pathways were the most prominent pathways. This was followed by 4 cancer pathways and the MAPK signaling pathway (Figure 4e). These results demonstrate that ATP5A1 regulates cancer cell activity by regulating alternative splicing of glucose homeostasis and cancer-related genes.

Alternative splicing analysis by ATP5A1-OE. (a) Bar plot showing the ratio of spliced reads in ATP5A1 and control samples (Student's t-test, * P < 0.05). (b) Bar plot showing the number of RASEs by classifying them into ten types. (c) Venn diagram showing the overlapping genes between the DEGs and RASGs. (d) Bubble plot showing the top ten enriched GO BP terms for RASGs. Terms related to cancer pathways are labeled in red. (e) Bubble plot showing the top ten enriched KEGG pathways for RASGs. Terms related to cancer pathways are labeled in red.
Validation of the ATP5A1-Regulated Alternative Splicing Events
From the above results, it was noted that the enriched KEGG pathways associated with RASGs were linked to multiple cancer pathways. Several RASEs were then selected from pathways that included the MAPK, Notch, and mTOR signaling pathways, as well as a pathway associated with cancer. RT-qPCR experiments were then performed to validate the significant differences, including 11 non-intron retention (non-IR) events and 3 IR events, observed in HeLa cells. The RASEs that were tested included the MAP4K3, MAPK9, RPTOR, NCOR2, APH1B, STRADA, DUSP16, STAT3, PDPK1, MKNK2, MAP4K4, EGFR, DLL3, and MSH2 genes. As described in the methods, specific primers were designed for each RASE, and the ratio between the modeled and alternative events is calculated. From the tested RASEs, a high consistency between RNA-seq and RT-qPCR results was found. 7 RASEs, which included DUSP16, NCOR2, MKNK2, PDPK1, and STAT3, were validated and found to have a significant change in the ratio between ATP5A1-OE and control samples (Figure 5, Figure S3). These results demonstrated the important functions of ATP5A1 in AS regulation in HeLa cells.

RT-qPCR validation of RASEs from cancer-related pathways. (a) to (c) Validation of 3 RASEs in HeLa cells. The number of junction reads was marked on the line representing RASE-associated splice junctions. The structures of the RASEs are depicted in the top-right panel. The altered RASE ratios between RNA-sequencing and in RT-qPCR results were calculated and plotted (right panel, bottom).
Discussion
In this study, the ATP5A1-regulated transcriptome profile was extensively investigated in HeLa cells for the first time. It is well established that ATP5A1 participates in the activity of ATP synthase in mitochondria. Recent studies reported that abnormal ATP5A1 expression is associated with the development of several cancers, including clear cell renal cell carcinoma 30 and glioblastomas. 31 However, the underlying molecular mechanisms were not clear. Inspired by the discovery that, using the interactome capture method, ATP5A1 has the ability to interact with RNA, 18 ATP5A1 was overexpressed in HeLa cells and whole transcriptome sequencing was performed to explore its effects on gene expression and alternative splicing. It was found that ATP5A1 interfered with the global transcriptome profile and induced 589 DEGs and 572 RASGs. Functional analysis of these dysregulated genes demonstrated that they were associated with multiple aspects of cancer development. This included angiogenesis, the collagen catabolic process, glucose homeostasis, and cellular responses to hypoxia.
In previous studies, ATP5A1 was identified and recognized as a subunit of mitochondrial ATP synthase.22,48 The downregulation of mitochondrial H+-ATP synthase enables cancer cells to reprogram energy metabolism to support their growth and progression and has been observed in several human carcinomas.49,50 However, it is not clear what the cellular outcome of ATP synthase inhibition in cancer cells is. Suggesting its importance in biological functions, reduced levels of ATP5A1 correlate with earlier-onset prostate cancer. 51 Our finding that overexpression of ATP5A1 promotes cellular apoptosis suggests that ATP5A1 could inhibit cancer progression by inducing the apoptosis pathway in cancer cells. To decipher the underlying mechanisms, the DEGs caused by ATP5A1 overexpression in HeLa cells were explored. The collagen catabolic process was the GO BP term most enriched across the upregulated genes. Collagen is the most prevalent component of the extracellular matrix (ECM) and plays an important role in tissue architecture and reprogramming. 52 It has been demonstrated that the metabolism and reconstruction of collagen are tightly associated with cancer development and progression. Collagen prolyl hydroxylases have the ability to promote the alignment of breast cancer cells on collagen fibers thereby enhancing their invasion and metastasis to the lymph nodes and lungs. 53 Collagen VI can directly affect malignant cells by acting on the Akt–GSK-3β–β-catenin–TCF/LEF axis, thus enhancing the production of protumorigenic factors and inducing epithelial–mesenchymal transition. 54 The metabolism regulatory functions of proline, which can be derived from collagen degradation, plays a role in apoptosis, autophagy, and nutrient metabolism in cancer. 55 The 5 genes (MMP3, MMP2, MMP19, KLK6, and ADAMTS2) we found to be upregulated by ATP5A1 all participate in the collagen catabolic process. This suggests that the induced apoptosis in HeLa cells by ATP5A1 may be caused by the elevated expression of MMP proteins.
The second most enriched term in the upregulated genes was associated with angiogenesis. Pathological angiogenesis is a hallmark of cancer and other diseases.56,57 Previous studies have demonstrated that matrix petallopeptidases (MMPs), especially MMP19, are tightly associated with angiogenesis in tumor tissue.58–60 Enriched in the angiogenesis pathway, MMP2 and MMP19 were found to be upregulated by ATP5A1 in this study. In MMP19−/− mice, earlier onset of tumoral angiogenesis and increased tumor invasion were observed, suggesting that MMP19 is a negative regulator of the early steps of tumor angiogenesis and invasion. 61 Similarly, a separate study also validated the tumor suppressor and anti-angiogenic activities of MMP19 in nasopharyngeal carcinoma. 62 MM2 is another matrix petallopeptidase associated with angiogenesis and has been found to promote lung cancer angiogenesis by altering VEGF expression. 63 Polymorphisms in the angiogenesis-related genes, including MMP2, are associated with the survival of colorectal cancer patients. 64 However, MMP2 had a very low expression level in HeLa cells in this study (FPKM <0.1), suggesting that it has a limited role in cervical cancers. The expression level of MMP19 was much higher than MMP2, suggesting that MMP19 plays a more dominant role in the angiogenesis pathway in our data.
In this study, the expression of several heat shock genes associated with the 70 kDa protein family, including HSPA1A, HSPA6, and HSPA1B, were greatly increased in ATP5A1-OE samples. Numerous studies have demonstrated their important regulatory functions and their therapeutic potential in multiple cancers.65–67 HSP70 knockdown by siRNA could significantly increase apoptosis level in HSC, MCF-7, and Huh7.5 cells after plasmonic photothermal therapy. 68 1 study showed that HSP70 to 2 is expressed in cervical carcinoma and it is involved in the growth, migration, and invasion of cervical cancer cells. 69 An HSP70 vaccine was used in combination with a soluble B and T lymphocyte attenuator in a murine TC-1 cervical cancer model. 70 However, it has not been validated whether HSP70 could inhibit cellular apoptosis in cervical cancer cells. In our study, the expression of HSP70 protein-associated RNAs was highly elevated by ATP5A1-OE, suggesting that HSP70 chaperone proteins may be activated and play important roles in cervical cancer cells. Further studies are needed to decipher the molecular and cellular functions, including apoptotic regulation, of activated HSP70 proteins.
Another interesting finding in this study involved the post-transcriptional regulation of ATP5A1, particularly, with regard to the regulation of alternative splicing. Cassette exon events were specifically repressed in overexpressed ATP5A1 samples. Alternative splicing changes the splicing pattern of specific isoforms of numerous genes and is tightly associated with cancer. 47 We found that the ATP5A1-regulated AS genes were enriched in multiple cancer-associated processes, including glucose homeostasis and cellular responses to hypoxia. KEGG pathway analysis revealed that the HIF-1 and mTOR signaling pathways were also enriched. The AS-associated dysregulation of genes in the HIF-1 signaling pathway indicates that ATP5A1 has the ability to modulate HeLa cells in response to hypoxia. Overexpression of ATP5A1 may alter the homeostasis of oxidative phosphorylation and energy metabolic pathways in cancer cells. HIF-1 signaling activation plays a key role in the reprogramming of cancer metabolism by activating the transcription of genes encoding glucose transporters and glycolytic enzymes, 71 while HIF-1 activation by hypoxia is a key regulator of angiogenesis in cancer.72,73 We have found that angiogenesis-related genes were upregulated by ATP5A1 (Figure 3a). We, therefore, hypothesize that AS modulation by ATP5A1 is another important regulatory mechanism in response to the reprogrammed energy metabolism.
Further studies are needed to make a thorough investigation of the underlying molecular mechanisms, especially as it relates to how ATP5A1 regulates gene expression and alternative splicing by acting as an RNA binding protein. 1 limitation of this study is that we only used HeLa cells to perform these experiments. Although HeLa cells are widely used in tumor and molecular biology studies, our findings will be more conclusive if the discoveries are validated in other cell lines. While an in-depth exploration on how ATP5A1 promotes cellular apoptosis in HeLa cells was also not made, ATP5A1-induced apoptosis may likely be facilitated through the mitochondrial apoptotic pathway. As only the expression levels of genes from BCL-2 family members were analyzed, further studies are needed to establish a full understanding of the underlying molecular mechanisms.
In summary, we have successfully used RNA-seq technology to demonstrate the changes in the transcriptome profile due to the overexpression of ATP5A1 in HeLa cells. These results demonstrate that ATP5A1 participates in important functional pathways involved in cellular apoptosis, response to hypoxia, and energy metabolism. ATP5A1 regulation of these pathways is enforced not only by altered expression levels but also by modulating alternative splicing of pathway-associated genes. Our results extensively broaden our understanding of the functions of ATP5A1 in cancer cells and provide a novel insight into the functional manner in which ATP5A1 regulates biological processes related to tumor development.
Supplemental Material
sj-docx-1-tct-10.1177_15330338211039126 - Supplemental material for ATP5A1 Participates in Transcriptional and Posttranscriptional Regulation of Cancer-Associated Genes by Modulating Their Expression and Alternative Splicing Profiles in HeLa Cells
Supplemental material, sj-docx-1-tct-10.1177_15330338211039126 for ATP5A1 Participates in Transcriptional and Posttranscriptional Regulation of Cancer-Associated Genes by Modulating Their Expression and Alternative Splicing Profiles in HeLa Cells by Yisa Song, BA, Fei Wang, MA, Yaxun Wei, MA, Dong Chen, BA, Gang Deng and BA in Technology in Cancer Research & Treatment
Supplemental Material
sj-xlsx-2-tct-10.1177_15330338211039126 - Supplemental material for ATP5A1 Participates in Transcriptional and Posttranscriptional Regulation of Cancer-Associated Genes by Modulating Their Expression and Alternative Splicing Profiles in HeLa Cells
Supplemental material, sj-xlsx-2-tct-10.1177_15330338211039126 for ATP5A1 Participates in Transcriptional and Posttranscriptional Regulation of Cancer-Associated Genes by Modulating Their Expression and Alternative Splicing Profiles in HeLa Cells by Yisa Song, BA, Fei Wang, MA, Yaxun Wei, MA, Dong Chen, BA, Gang Deng and BA in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Acknowledgments
We are very thankful to Dr Yi Zhang's team members for their helpful discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Scientific research project of Qinghai Health and Family Planning Commission (Grant Number 2017-wjzdx-04) and Young doctor support program of ABLife Inc., Wuhan (Grant Number 7702419).
Ethics Approval
Not applicable, HeLa cell line was purchased from CCTCC (China Center for Type Culture Collection, Wuhan, Hubei, China) and did not require ethics approval for their use in this study.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.