
Abstract
Background:
Dysregulation of microRNAs (miRNAs) in papillary thyroid cancer (PTC) might influence prognosis of PTC. This study is aimed to develop a risk score system for predicting prognosis of PTC.
Methods:
The miRNA and gene expression profiles of PTC were obtained from The Cancer Genome Atlas database. PTC samples were randomly separated into training set (n = 248) and validation set (n = 248). The differentially expressed miRNAs (DE-miRNAs) in the training set were screened using limma package. The independent prognosis-associated DE-miRNAs were identified for building a risk score system. Risk score of PTC samples in the training set was calculated and samples were divided into high risk group and low risk group. Kaplan-Meier curves and receiver operating characteristic (ROC) curve were used to assess the accuracy of the risk score system in the training set, validation set and entire set. Finally, a miRNA-gene regulatory network was visualized by Cytoscape software, followed by enrichment analysis.
Results:
Totally, 162 DE-miRNAs between tumor and control groups in the training set were identified. An 8 independent prognosis-associated DE-miRNAs, (including miR-1179, miR-133b, miR-3194, miR-3912, miR-548j, miR-6720, miR-6734, and miR-6843) based risk score system was developed. The area under ROC curve in the training set, validation set and entire set was all above 0.93. A miRNA-gene regulatory network involving the 8 DE-miRNAs were built and functional enrichment analysis suggested the genes in the network were significantly enriched into 13 pathways, including calcium signaling pathway and hedgehog signaling pathway.
Conclusion:
The risk score system developed this study might be used for predicting the prognosis of PTC. Besides, the 8 miRNAs might affect the prognosis of PTC via hedgehog signaling pathway and calcium signaling pathway.
Keywords
Introduction
Thyroid cancer (TC) is a popular endocrine tumor, which is mainly divided into papillary thyroid cancer (PTC), medullary thyroid cancer (MTC), follicular thyroid cancer (FTC), and anaplastic thyroid cancer (ATC). 1 TC is mainly caused by enlarged thyroid, family history, and radiation exposure. 2 It usually has the symptoms of neck swelling and neck lump, 3 and is more common in young females. 4 Globally, TC affects the life of 3.2 million people and is responsible for 31,900 deaths in 2015. 5 Especially, PTC is the most predominant form, accounting for 75% to 85% of TCs. 6 Therefore, the mechanisms of PTC should be further researched.
MicroRNAs (miRNAs) are a kind of short non-coding RNAs, which can regulate the expression levels of genes involved in multiple biological processes. 7,8 Several miRNAs have been reported to play important role in PTC and have potential to be biomarkers or therapy targets for PTC. For example, miR-335 expression is reduced in PTC samples and could repress cell growth and metastasis in PTC by mediating zinc finger E-box binding homeobox 2 (ZEB2), therefore, miR-335 can serve as a tumor suppressor in PTC. 9 Through regulating fibronectin 1 (FN1), miR-139 acts as a tumor suppressor and represses the tumor formation in PTC. 10 MiR-451 is a valuable tissue marker for PTC, which may also be a noninvasive blood marker for diagnosing and evaluating metastasis of PTC. 11 MiR-144-3p facilitates the cell growth and metastasis in PTC through mediating paired box gene 8 (PAX8), which may be useful prognostic marker and promising therapeutic target for PTC. 12,13 However, the accuracy of single miRNA for predicting prognosis of PTC is usually not ideal.
In 2019, Pak et al built a gene expression-based risk score system for predicting the event-free survival and the presence of metastasis in PTC patients. 14 Nevertheless, the miRNA expression-based risk score system for predicting the overall survival of PTC patients has not been established. Combined with the miRNA expression data of PTC samples in The Cancer Genome Atlas (TCGA) database, the current study screened the independent prognosis-associated miRNAs in PTC for constructing the risk score system. Combined with the mRNA expression profile of PTC samples, the functions of the independent prognosis-associated miRNAs were analyzed. Our findings might help to reveal the action mechanisms of key miRNAs in PTC and predict the prognosis of patients with PTC.
Materials and Methods
Data Source
The miRNA expression data (including 568 samples) and mRNA expression data (including 573 samples) of PTC (platform: Illumina HiSeq 2000 RNA Sequencing; downloaded in June 15, 2019) were obtained from TCGA database (https://cancergenome.nih.gov/). After matching with the clinical information of the samples, a total of 553 samples (including 57 normal controls and 496 PTC samples) with both miRNA data, mRNA expression data and survival prognosis information were selected. The age of the PTC patients were 47.20 ± 15.99 years old. A total of 299 patients received radiation therapy, 178 patients didn’t receive radiation therapy and the radiation therapy of 19 patients was not available. The overall survival time was 42.20 ± 34.58 years old. The 496 PTC samples (the entire set) were randomly and equally divided into 2 groups (training set: 248 samples; and validation set: 248 samples). The clinical information of the entire set, the training set, and the validation set were arranged and listed in Table 1.
The Clinical Information of the Training Set, the Validation Set and the Entire Set.

Note: SD, standard deviation.
Differential Expression Analysis
The samples were classified into tumor and control groups according to their source information. Using the limma package (version 3.34.7, https://bioconductor.org/packages/release/bioc/html/limma.html) 15 in R, the differentially expressed miRNAs (DE-miRNAs) between tumor and control groups were screened. The resulting P value was adjusted by Benjamini & Hochberg method to reduce false discovery rate (FDR). DE-miRNAs were selected based on FDR and |log2 fold change (FC)|. FDR < 0.05 was set and |log2 (FC)| was adjusted to obtain enough number of DE-miRNAs for further analysis. At last, FDR < 0.05 and |log2 (FC)| > 0.5 were used as the thresholds for screening DE-miRNAs. Combined with the expression values of the DE-miRNAs in the training set, hierarchical clustering was conducted using the pheatmap package (version 1.0.8, https://cran.r-project.org/web/packages/pheatmap/index.html) 16 in R.
Establishment of Risk Score System
Combined with the prognosis information of the 248 PTC samples in the training set, the DE-miRNAs significantly related to overall survival were selected using the univariate Cox regression analysis in the survival package (version 2.41 -1, http://bioconductor.org/packages/survivalr/)17 in R. The log-rank p-value < 0.05 was taken as the threshold.
Using the multivariate Cox regression analysis in the survival package, 17 the DE-miRNAs significantly correlated with independent prognosis were further identified. Based on the independent prognosis-associated DE-miRNAs, the risk score system was established using following formula:
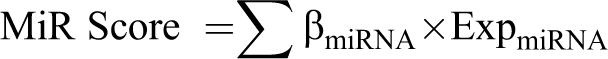
Where β miRNA stands for the independent prognostic coefficient of independent prognosis-associated DE-miRNA, and Exp miRNA represents the expression level of independent prognosis-associated DE-miRNA.
The risk score of each sample in the training set was calculated. Then, the samples in the training set were divided into high risk group and low risk group on the basis of the median value of the risk score. Kaplan-Meier (KM) curve in the survival package17 was draw for samples in high risk group and low risk group. The significance of the overall survival difference between high risk group and low risk group was calculated with log-rank test.
Validation of risk score system in validation set and entire set
Based on the risk score system established in the training set, the PTC samples in the validation set and the entire set were divided into high risk group and low risk group. KM method and log-rank test were used to test the efficiency of the risk score system in the the validation set and the entire set.
Screening of Independent Prognostic Factors
Using the univariate Cox regression analysis in the survival package, 17 the clinical factors significantly correlated with overall survival were screened from the training set, the validation set and the entire set, respectively. To identify the independent clinical prognostic factors, the screened clinical factors were further subjected to multivariate Cox regression analysis. 18 The threshold was set as log-rank p-value < 0.05.
Subsequently, stratification analysis was conducted for the independent clinical prognostic factors to reveal their correlations with risk grouping. Risk stratification analysis of the training set, the validation set and the entire set was respectively conducted to study the prognosis conditions of the independent clinical prognostic factors in different risk groups.
Regulatory Network and Functional Enrichment Analysis
The differentially expressed genes (DEGs) between high and low risk groups of the entire set were analyzed using the limma package. 15 The |log2 FC| > 0.263 and FDR < 0.05 were set as the thresholds for screening DEGs. Moreover, hierarchical clustering for the DEGs was performed with the pheatmap package. 16
The starBase database (version 2.0, http://starbase.sysu.edu.cn/) 19 included the target gene prediction information in RNA22, PITA, targetScan, picTar, and miRanda databases. Using starBase database, the targets of the independent prognosis-associated DE-miRNAs were searched. The miRNA-gene regulatory relationships predicted by 2 or more databases of RNA22, PITA, targetScan, picTar, and miRanda were selected. Then, the DEGs were mapped to the target genes of the independent prognosis-associated DE-miRNAs. The overlapped miRNA-DEG relationship were filtered to build miRNA-gene regulatory network using Cytoscape software (version 3.6.1, https://cytoscape.org/). 20 Further, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analyses were carried out for the genes involved in the regulatory network. Significant pathways and GO terms were selected with p-value < 0.05 as the threshold.
Results
Differential Expression Analysis of Samples in Training Set
After tumor (248 samples in the training set) and control (57 normal control samples) groups were divided, 162 DE-miRNAs between the 2 groups were screened, including 59 up-regulated miRNAs and 103 down-regulated miRNAs. The scatter diagram and clustering heatmap based on the expression levels of the DE-miRNAs are presented in Figure 1A and B, respectively. From Figure 1B, the expression patterns of DE-miRNAs were obvious different between PTC samples and control samples.

The scatter diagram and bidirectional hierarchical clustering heatmap based on the expression levels of the differentially expressed miRNAs (DE-miRNAs). (A) The scatter diagram (the horizontal and vertical axises represent the expression levels of control group and tumor group, respectively; red, green, and blue dots separately represent up-regulated miRNAs, down-regulated miRNAs and non-DE-miRNAs); (B) The bidirectional hierarchical clustering heatmap (in sample strip, black and white separately represent tumor samples and control samples).
Construction of Risk Score System
From the 248 PTC samples in the training set, 20 DE-miRNAs significantly correlated with overall survival were identified. Based on multivariate Cox regression analysis, 8 independent prognosis-associated DE-miRNAs, including miR-1179, miR-133b, miR-3194, miR-3912, miR-548j, miR-6720, miR-6734, and miR-6843, were further screened (Table 2). Afterward, the risk score system based on the independent prognosis-associated DE-miRNAs was constructed. The calculation formula was:
The Differentially Expressed miRNAs (DE-miRNAs) Significantly Correlated With Independent Prognosis.

Note: CI, confidence interval.

The risk score of each sample in the training set were calculated and the PTC samples were divided into high risk group and low risk group. The difference of overall survival in high risk group and low risk group was calculated. As shown in Figure 2A, the overall survival rate of samples in the low risk group was significant longer than that in the high risk group (P < 0.05). The area under the ROC curve (AUC) of 3-year prediction and 5-year prediction was both 0.998, indicating higher accuracy and credibility of the risk score model.

The distribution diagram of risk scores (RSs), the Kaplan-Meier (KM) curves and receiver operating characteristic (ROC) curves for the training set (A), validation set (B) and entire set (C). In RS distribution diagrams, the horizontal and vertical axises separately represent sample numbers and RSs. In KM curves, blue and red curves separately indicate low risk and high risk. In ROC curves, red and black curves separately represent 3-year prediction and 5-year prediction. HR: hazard ratio; AUC, area under the receiver operating characteristic curve.
Validation of the risk score system in the validation set and entire set
The credibility of the risk score system was further validated in the validation set and the entire set, respectively. The overall survival rate of samples in the low risk group was significantly longer than that in the high risk group in both validation set (Figure 2B) and entire set (Figure 2C) (P < 0.05). The 3-year prediction AUC and 5-year prediction AUC in both validation set and entire set were larger than 0.93.
Screening of independent prognostic factors
The association between clinical information and overall survival was analyzed using univariate Cox regression analysis and multivariate Cox regression analysis in the training set, validation set and entire set, respectively. Age and MiR Score status were identified as independent clinical prognostic factors in the training set, the validation set and the entire set, respectively (Table 3). In the entire set, the training set, and the validation set, the PTC patients with lower age had better survival prognosis (Figure 3). These results were consistent with the actual situation. Furthermore, stratification analysis was conducted for age to reveal its correlation with risk grouping in the training set, the validation set and the entire set. KM curves suggested that age was significantly related to overall survival in the high risk group (Figure 4).
The Independent Clinical Prognostic Factors Selected From the Training Set, the Validation Set and the Entire Set.

Note: HR, Hazard Ratio; CI, confidence interval.

The Kaplan-Meier (KM) curves of different age groups in the training set (A), the validation set (B) and entire set (C). Blue and red curves indicate age ≤ 50 years and age ≥ 50 years, respectively.

The Kaplan-Meier (KM) curves showing the correlations between age in different risk groups and overall survival. (A) The KM curves for the low risk group (left: the training set; middle: the validation set; and right: the entire set). (B) The KM curves for the high risk group (left: the training set; middle: the validation set; and right: the entire set). Blue and red curves represent the samples no more than 50 years and the samples older than 50 years, respectively. HR, hazard ratio.
Regulatory Network and Functional Enrichment Analysis
Total 469 DEGs, including 427 up-regulated genes and 42 down-regulated genes were identified between the high risk group and low risk group of the entire set (Figure 5A). The expression patterns of DEGs changed with MiR Score is presented in Figure 6B.

The volcano plot and heatmap for the differentially expressed genes (DEGs). (A) The volcano plot (the horizontal dashed line indicates false discovery rate (FDR) < 0.05, and the vertical dashed lines represent |log2 fold change (FC)| > 0.263; the blue dots stand for the DEGs); (B) The heatmap based on the expression levels of the DEGs (left-to-right order indicates the changes of risk scores from low to high).

The miRNA-gene regulatory network. Circles and squares represent mRNAs and miRNAs, respectively. FC, fold change.
The target genes of the 8 independent prognosis-associated DE-miRNAs were predicted in starBase version 2.0. The obtained target genes were compared with the 469 DEGs, resulting in 176 overlapped DEGs. A miRNA-DEG regulatory network was constructed, involving 8 DE-miRNAs, 176 DEGs and their 515 interactions (Figure 6).
Functional enrichment analysis revealed that the genes in the regulatory network were significantly enriched into 17 significant GO_ Biology Process (BP) terms, such as “regulation of system process” (p-value = 1.36E-04), “regulation of cell proliferation” (p-value = 9.06E-04) and “transmission of nerve impulse” (p-value = 1.50E-03) and 13 KEGG pathways, such as “T cell receptor signaling pathway” (p-value = 1.15E-02), “hedgehog signaling pathway” (p-value = 1.26E-02) and “calcium signaling pathway” (p-value = 1.28E-02) (Table 4).
The Enrichment Results for the Genes Involved in the Regulatory Network.

Discussion
In the present study, 162 DE-miRNAs between tumor and control groups were screened. After 20 DE-miRNAs that significantly correlated with overall survival were identified, 8 independent prognosis-associated DE-miRNAs, including miR-1179, miR-133b, miR-3194, miR-3912, miR-548j, miR-6720, miR-6734, and miR-6843 were further selected for constructing the risk score system. KM curves and ROC curves suggested that the risk score system could predict the prognosis of PTC with high accuracy and credibility. Based on Cox regression analysis, age and MiR Score status were screened as the independent clinical prognostic factors There were 469 DEGs (427 up-regulated and 42 down-regulated) between the high risk group and low risk group of the entire set. A miRNA-DEG regulatory network was constructed and functional enrichment analysis suggested the genes involved in the regulatory network were significantly enriched in 17 GO_BP terms and 13 KEGG pathways.
The 8 DE-miRNAs, including miR-1179, miR-133b, miR-3194, miR-3912, miR-548j, miR-6720, miR-6734 and miR-6843 were identified as independent prognosis factors with PTC. Some of these miRNAs were reported to play important roles in PTC previously. MiR-1179 was one of the most downregulated molecules in samples with a papillary pattern of growth. 21 The downregulation of miR-1179 in PTC could lead to the deregulation of cell cycle progression or invasiveness. 21 MiRNA-1179 was also identified in PTC samples using first next-generation sequencing study. 22 Eleven dysregulated miRNAs (including down-regulated miR 1179) are identified in PTC tissues in comparison to normal thyroid tissue, which are related to different histological types and recurrence risks of PTC. 23
Though other 7 miRNAs were found differentially expressed in PTC and were found to be independently associated with prognosis of PTC, few studies have conducted to investigate their role in PTC. However, their roles in other cancers were reported. MiR-3194-5p may be candidate targets for blocking the metastasis of the oropharyngeal squamous cell carcinoma development. 24 MiR-3912 and miR-548j expression in embryonal tumors and the controls are differential, which may possess diagnostic value for Atypical Teratoid/Rabdoid Tumors (AT/RTs). 25 MiR-548-3p suppresses the cell proliferation in breast cancer via mediating enoyl-CoA hydratase, short chain 1 (ECHS1), which may be a candidate target for the treatment of the tumor. 26 The miR-548e-5p-ubiquitin conjugating enzyme E2 C (UBE2C)-ZEB1/2 axis can promote cell invasion in lung cancer, and may be valuable for diagnosing and treating lung cancer. 27 MiR-133b can significantly suppress cell proliferation, metastasis, and apoptosis in colorectal cancer (CRC) through the TAp63/miR-133b/ras homolog family member A (RhoA) loop. 28 MiR-133b dysregulation may be correlated with TAp63 down-regulation in CRC, and miR-133b and TAp63 may be effective prognostic markers for the disease. 29 MiR-9-5p, miR-135a-5p, miR-129-5p, and miR-6720-3p may mediate pathways in cancer, and miR-6720 expression is elevated in hepatocellular carcinoma patients treated by alternariol. 30,31 MiR-6734 suppresses colon cancer cell growth via increasing p21 levels, therefore, miR-6734 exerts a key regulatory effect on tumor cell proliferation and survival. 32 Thus, these 7 miRNAs might play important roles in PTC and are warrant to be investigated in PTC.
Previous studies have developed other miRNA-based signatures using TCGA data for predicting prognosis of PTC. For example, a 2-miRNA signature (hsa-miR-181a-2-3p and hsa-miR-138-1-3p) was identified by Liu et al and the AUC of this 2-miRNA signature in predicting 5-year survival was 0.784. 33 Xiong et al developed a 4 miRNA signature for predicting survival of patients with PTC with an AUC of 0.886 for 5-year disease survival. 34 The AUC of the risk score system developed in this study for predicting 3-year and 5-year survival was all above 0.93 in the training set, validation set and entire set, which is higher than the miRNA signatures developed in previous study. Besides, the risk score system in this study only included 8 miRNAs. The cost of detecting the expression of these 8 miRNAs by quantitative Real-time PCR is about a few dollars to tens of dollars in different countries. Therefore, this risk score system might be a cost-effective method for predicting the prognosis of PTC patients.
The genes in the miRNA-DEG regulatory network were significantly enriched in 17 GO_ BP terms, such as “regulation of system process” (p-value = 1.36E-04), “regulation of cell proliferation” and 13 KEGG pathways, such as “T cell receptor signaling pathway,” “hedgehog signaling pathway” and “calcium signaling pathway.” These function terms are the canonical BPs or pathways that were dysregulated in cancer. Through activating c-Met and AKT, the sonic hedgehog pathway promotes cell motility and invasion in ATC. 35,36 In male rats, calcium signaling is implicated in the mode of action for TC induced by acrylamide exposure. 37 Hedgehog signaling pathway and calcium signaling pathway were enriched for the genes implicated in the regulatory network, indicating that the activation or suppression of the pathways might function in the prognosis of PTC.
Though the risk score system developed in this study is of high sensitively and specificity for predicting the prognosis of PTC patients in TCGA databases, there are some inevitable limitations in this study. First, the external validation of the risk score system is not conducted because there is no sufficient dataset of PTC with complete clinical information. Second, the validation in clinical practice is also warranted in the future study. We hypothesized this study, if confirmed, could significantly improve the prognosis predicting accuracy of PTC patients and gene therapy targeting these 8 miRNAs might improve the prognosis of these patients.
Conclusion
In conclusion, 162 DE-miRNAs between tumor and control groups were screened. Besides, an 8 miRNAs, including miR-1179, miR-133b, miR-3194, miR-3912, miR-548j, miR-6720, miR-6734 and miR-6843 risk score system was developed and this risk score is effective for predicting the prognosis of PTC. Furthermore, a miRNA-DEG regulatory network was constructed based on the 8 independent prognosis-associated DE-miRNAs and their target DEGs. Functional enrichment analysis suggested that these DEGs were significantly enriched in 13 pathways, including hedgehog signaling pathway and calcium signaling pathway. However, more experimental studies should be conducted in the follow-up study to confirm our results.
Footnotes
Authors’ Note
This paper does not include human or animal studies, so the ethics statement is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by “the Fundamental Research Funds for the Central Universities” Project No. 22120180392 and 15012150066. Supported by “Shanghai Tenth People’s Hospital” Project No. 04.03.18.105.Supported by “Shanghai Science and Technology Commission Support Plan” Project No.18441903500.Supported by “Shanghai Outstanding Academic Leader program” Project No. 18XD1403000.Shanghai Leading Talent program sponsored by Shanghai Human Resources and Social Security Bureau with number 03.05.19005;