
Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the most aggressive cancer types in China. In recent years, progress has been made in various types of cancer genomics including ESCC. However, the clinical significance of genomic variation of ESCC remains poorly defined. In the present study, genomic sequencing data from 469 ESCC cases were analyzed and potential therapeutic targets in the Druggable Genome Interaction Database (DGIdb) were screened. A series of potential therapeutic target genes and pathways were identified, of which treatment of ESCC with bortezomib (a specific inhibitor targeting proteasome) potently inhibited the proliferation of 5 ESCC cell lines and administration of bortezomib led to significant tumor xenograft regression in SCID mice. It was also identified that kinase insert domain receptor (KDR), which had drug recommendations from all 6 sources integrated by the DGldb and harbored significant amplification in ESCC, might be a downstream target of zinc finger protein 750 (ZNF750). ZNF750 acts as a transcription factor and has been demonstrated to harbor frequently inactivating mutations in ESCC by previous independent studies. In the present study, KDR was upregulated upon ZNF750 knockdown and the rescue of ZNF750 also led to marked restoration of KDR. KDR knockdown in stable ZNF750-knockdown KYSE150 and KYSE140 ESCC cells significantly attenuated the promotion of cell growth, colony formation, invasion and migration induced by ZNF750 knockdown. Further experiments found that apatinib treatment, a potent inhibitor of KDR, resulted in profound inhibition of cell proliferation and invasion. Collectively, the present study provided insight for genomic alterations as potential therapeutic targets in ESCC and supported the possibility of a therapeutic strategy targeting the proteasome in ESCC. The present results also suggested that targeting KDR may be an effective way to treat ESCC, not only in KDR variant cases, but also in individuals with ZNF750 mutations and deletions.
Introduction
Esophageal cancer is one of the most lethal cancers worldwide and globally. More than half of esophageal cancers occur in China, and the main histologic type in China is esophageal squamous cell carcinoma (ESCC). 1 Due to the limitation of early diagnosis and treatment method, the 5-year survival rate of ESCC ranges between 10 and 25%. 2 Different from successful use of molecular targeted therapy for other solid tumors, molecular targeted therapy for ESCC is still immature and known targets are very limited.
Recently, much progress has been made in various types of cancer genomics, including in ESCC with the progression of next-generation genomic sequencing. 3 -7 These assays were conducted in our previous study, revealing genomic alterations and identifying a series of significantly mutated genes, which could help provide insight for selecting a suitable therapy for specific molecular abnormalities. Studying the gene-drug interaction based on genomic data and identifying an association between molecular variations and good therapeutic agents for cancer patients is a challenge for medical decision-making. As a result, some public resources have been developed to connect genomic molecular variation with appropriate clinical therapies. These resources included the Druggable Genome Interaction Database (DGIdb), Personalized Cancer Therapy and My Cancer Genome (MCG). Among them, The DGIdb integrates 15 disparate sources and serves as a useful and comprehensive tool for defining the genes or gene product analogues that are known to interact with drugs and/or are potentially druggable. 8
In our previous study, whole-genome sequencing (WGS) of 14 tumor and matched samples, and whole-exome sequencing (WES) of 90 ESCC and adjacent normal tissue from individuals in a high incidence area of China was performed. 3 Mutational signatures that cause somatic mutations in ESCC were described and driver genes contributing to tumorigenesis of ESCC were identified. In the present study, previously published genomic sequencing data of ESCC in the DGIdb database were analyzed, aiming to identify a series of candidate druggable genes in ESCC. ZNF750 as a key differentiation regulator in squamous epithelium has attracted increasing attention due to its significant mutations and frequent deletions in ESCC. 3,5 Our previous study demonstrated that ZNF750 mutations encoding truncated or disrupted proteins might contribute to ESCC tumorigenesis. 3 It has been demonstrated that ZNF750 acts as tumor suppressor in different ways in different cancer types. 9 -11 However, the downstream targets of ZNF750 and regulated genes in ESCC have not been studied extensively. In the present study, it was identified that the effect of ZNF750 on ESCC might be partly mediated by kinase insert domain receptor (KDR). KDR, also known as vascular endothelial growth factor receptor-2 (VEGFR-2), which plays an important role in tumor angiogenesis, was also one of the few genes with drug recommendations from all 6 sources integrated by The DGldb. It is well documented that KDR is markedly upregulated in many types of cancer cells 12 -14 and specific tyrosine kinase inhibitors (TKIs) of KDR, such as sorafenib and sunitinib, have significant antitumor effects. 15,16 Although inhibiting KDR is an important antitumor therapeutic target in many cancer types, KDR was seldom previously implicated in ESCC therapy.
The present study provided insight for genomic alterations as potential therapeutic options and screened DGIdb-associated druggable genes in ESCC. The present study revealed that inhibiting KDR might be a therapy option for patients with ESCC, especially harboring the ZNF750 inactivating variation. In addition, the present Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of all druggable genes in the DGIdb suggested that a proteasome inhibitor might be an effective treatment agent for ESCC therapy.
Materials and Methods
Samples and Genomic Data
Genome sequencing data of 469 ESCC patients from 5 independent cohorts were analyzed in the present study. Cohort 1 was derived from our previous study, which contained 104 ESCC cases. In this cohort, 14 tumors and matched normal samples underwent WGS and 90 samples underwent WES. 3 Cohort 2 was from a previous study by Song et al, which included 17 WGS and 71 WES ESCC samples (https://www.ebi.ac.uk/ega/studies/ EGAS00001000709). 4 Cohort 3 was from 20 WES samples from a previous study by Li et al (https://trace.ncbi.nlm.nih.gov/Traces/ sra/? study=SRP033394). 5 Cohort 4 was from a previous study by Gao et al, which contained 113 WES samples (http://www.ebi.ac.uk/ega/studies/EGAS 00001000932). 6 Cohort 5 was from 144 Japanese WES data from Sawada et al (http://dx.doi.org/10.1053/j.gastro. 2016.01.035). 7 None of ESCC individuals of 5 cohorts were treated by chemotherapy or radiotherapy before the sample collection. All the obtained variations in this study were somatic mutations, which is filtered by matched normal tissue or blood.
Web Tools and Pathway-Enrichment Data Analyses
The DGIdb database (http://dgidb.genome.wustl.edu) is affiliated with Washington University School of Medicine in St. Louis and incorporates 15 other known sources of drug-gene interactions. The DGIdb contains 26,298 particular drug-gene interactions, involving 2,644 genes, 7,569 drugs and 7,524 unique genes belonging to one or more of 41 potentially druggable gene categories. 8 The “search interactions” tool is for known drug-gene interactions. The pathway-enrichment analyses were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID; v.6.7) by examining the distribution of the druggable genes identified within KEGG.
Cell Culture
All the human ESCC cell lines in this study and HEK-293 cells were preserved in our laboratory. All the cell lines were inspected to be free of mycoplasma contamination. HEK-293 cell was used as a packaging cell line to produce virus. All ESCC cells were grown in DMEM/F12 media (Hyclone, Logan, UT, USA) supplemented with 10% FBS (Gibco, ThermoFisher Scientific), 100 units/ml penicillin and 100 μg/ml streptomycin. The cells were cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Knockdown and Overexpression of Genes in ESCC Lines
Lentivirus vector pLKO.1-puro and its packaging plasmids, pMD2.G and psPAX2, were obtained from Addgene, Inc. HEK-293 cells were transfected with the packaging plasmids along with the lentiviral small hairpin (sh)RNA vector using Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol. The medium was changed after 6 h. The viruses were harvested at 24 h post-transfection, passed through 0.22 µm filters, and freshly used for shRNA infection. The target ESCC cells (KYSE150 and KYSE140 with stable ZNF750 knockdown) were infected with lentiviral particles and incubated at 37°C for 24 h and then the viral supernatant was replaced with fresh media. After 48 h, the infected cell populations were selected using puromycin (3 μg/ml). After 5 days of selection, shRNA knockdown efficiency was determined by reverse transcription (RT)-PCR and western blot analysis. The shRNA sequences targeting ZNF750 were 5’-CCGGCTGCCGATTCCTTACGGATTTCTCGAGAAATCCGTAAGGAATCGGCAGTTTTTTG-3’; The shRNA sequences targeting KDR were 5’-CCGGCGCTGACATGTACGGTCTATGCTCGAGCATAGACCGTACATGTCAGCGTTTTTTG-3’ and 5’-CCGGGACTGGCTTTGGCCCAATAATCTCGAGATTATTGGGCCAAAGCCAGTCTTTTTTG-3’. For overexpression experiments of ZNF750 rescue, viruses were purchased from Cyagen Biosciences, Inc. The wild-type ZNF750 gene was cloned into the lentivirus expression vector to produce pLV[Exp]-mCherry/Neo-EF1A>HA/hZNF750, with multiplicity of infection values of 150.
Cancer PathwayFinder PCR Array
The Human Cancer Pathway Finder RT 2 Profiler PCR Array was used to observe the mRNA relative expression of known cancer-related genes. This array in a 96-well plate contains specific primers for 84 genes representing 6 biological pathways, 5 housekeeping genes with one well for genomic DNA control, 3 wells for RT controls and 3 wells for positive controls. After cDNA was synthesized by use of RT 2 First Strand kit, real-time PCR was performed using RT 2 Profiler PCR Arrays combined with RT2SYBR Green Master mixes. Expression of genes being analyzed was normalized with respect to housekeeping gene using the 2-ΔΔCq method for comparing relative expression (upregulation > 2.0 and downregulation < 0.5) using Analysis Web (http://www.SAB iosciences.com/pcrarraydataanalysis.php).
RT-Quantitative (q)PCR
Total RNA of ESCC cells was extracted using the RNeasy mini kit (Qiagen) and quantified with NanoDrop. In total, 1 mg total RNA was reverse transcribed using the iScript RT-PCR kit from Bio-Rad Laboratories, Inc. All qPCR reactions were performed in triplicate with an Applied Biosystems step one plus (Thermo Fisher Scientific, Inc.). Relative expressions of ZNF750 and KDR were determined by normalization to GAPDH using a standard curve method with 10 serial dilutions, according to the manufacturer’s protocol. Primers for ZNF750 were Forward (F):5’-TACATGCACCCCACAATCCC-3’ and Reverse (R):5’-GGTGAGGCAGGAAGTGTCTC-3’; primers for GAPDH were F:5’-AGGTCGGAGTCAACGGATTTG-3’ and R:5’-GTGATGGCATGGACTGTGGT-3’; and primers for KDR were F:5’-CACCACTCAAACGCTGACATGTA-3’ and R:5’-AAGAGTGCGCCAACGAGC-3’.
Western Blot Analysis
The analyzed cells were lysed as previously described. 3 Subsequently, the collected lysate was centrifuged at 4°C at 18,624xg for 15 min and protein concentration was determined by use of the Bradford method. In total, 50 µg protein was loaded and separated by SDS-PAGE and transferred onto Immobilon-P membranes using the iBlot apparatus (Invitrogen; Thermo Fisher Scientific, Inc.). After blocking in 5% fat-free milk, the membrane was incubated with the appropriate diluted primary antibody overnight at 4°C. The primary antibodies were used with the following dilution: Actin (Transgen Biotech Co., Ltd.; 1:5,000), ZNF750 (Abcam; 1:500) and KDR (Abcam; 1:200). Antibody binding was detected with horseradish-peroxidase-conjugated anti-mouse (Sigma-Aldrich; Merck KGaA) or anti-rabbit (Cell Signaling Technology, Inc.) antibodies for 1 h at room temperature and chemiluminescence was measured using a LAS4000 Device Chemiluminescence System (Beijing Sage Creation Science Co., Ltd.). Each experiment was repeated at least 3 times.
MTT Assay
Bortezomib (a proteasome inhibitor) was obtained from Selleck Chemicals. ESCC cells were seeded at 5 x 103 cells per well in a 96-well plate and incubated for 24, 48, 72 and 96 h. At the indicated time periods, 20 µl 5 mg/ml MTT (Invitrogen; Thermo Fisher Scientific, Inc.) solution was added to each well and analyzed cells were incubated for 4 h under normal culture conditions until crystals formed. Then, MTT solution was removed and 200 µl DMSO was added to dissolve the crystals. After shaking for 10 min, the color intensity was measured using a Microplate Reader (Bio-Rad Laboratories, Inc.) at 490 nm. Each experiment had 4 replicates and at least 3 independent experiments were carried out.
Colony Formation Assay
Cells were suspended using the corresponding complete medium and seeded at a density of 1 x 103 cells/well in 6-well plates on Day 0 and incubated in normal conditions for 10-15 days. Cells were fixed with 4% polyformaldehyde for 15 min and washed using 1X PBS 3 times. Then, cells were stained with 1% crystal violet solution and washed using 1X PBS 3 times. The experiments were performed in triplicate and the number of colonies containing >50 cells was microscopically counted to calculate the number of colonies.
Invasion Assay
In total, 2 x 105 control group and experimental group cells were suspended in 100 µl serum-free medium and seeded in the upper chamber of the insert with an 8-µm micro-porous membrane (Costar; Corning, Inc.) coated with Matrigel (BD Biosciences). The lower chamber was added by 600 µl medium containing 20% FBS. Plates were incubated at 37°C for 24 and 48 h, and cells above the surface of the membrane were carefully removed by use of a cotton swab. The invaded cells under the surface of the membrane were fixed with 4% paraformaldehyde and stained with 1% crystal violet for 15-20 min. After washing with 1X PBS, the number of invaded cells was counted by fluorescence microscopy. A total of 4 fields were randomly chosen, and the invasion assays were repeated 3 times.
Migration Assay
The scratch assay was used to determine the migratory capacity of the cells. Analyzed cells were suspended in corresponding complete medium and seeded in 6-well plates. When cells reached 95-100% density, scratch wounds were made across the monolayer ESCC cells in each well with a 200-µl pipette tip and the non-adherent cells were washed off using medium. Fresh serum-free medium was added to the wells and inverted microscope images were captured at 0, 24, 48, 72 and 96 h. All experiments were conducted independently in triplicate.
In Vivo Experiments
The use of experimental animals strictly followed the guideline for tumor induction in mice and rats. Twenty four-week-old female athymic BALB/c nude mice weighing 19-22 g were obtained from the Shanghai Genechem Co., LTD. All mice were kept in standard, pathogen-free conditions under a 12-h light/dark cycle with 22-26°C temperature and 45-65% humidity and free access to food and water. The mice were injected subcutaneously in the right oxter region with Eca-109 ESCC cells at a density of 4 × 106 in 100 µl serum-free medium. Palpable tumors (approx.∼5 mm in diameter/mouse) were produced after 11 days, then the drug administration study was initiated (Day 1). The BALB/c nude mice were assigned randomly to 2 experimental groups with 10 mice in each group: The 200 µl normal saline group and the bortezomib group with 1.5 mg/kg bortezomib (in 200 µl normal saline) administered by intraperitoneal injection twice weekly. Eight hours after the final drug injection at week 3, the mice were euthanized by excessive injection of 2% sodium pentobarbital. The tumors were removed and measured with calipers. The volume of each tumor was calculated using the following formula, tumor volume = π/6×L×W×W. L represents length and W represents width.
Immunohistochemistry
Sections of nude mouse transplanted tumor tissue were incubated with specific antibodies at a 1:200 dilution for 14 h at 4°C, followed by detection using the PV8000 (Zhongshan, China) and a DAB detection kit (Maixin, China), producing a dark brown precipitate. Slides were counterstained with hematoxylin.
Statistical Analysis
SPSS 18.0 software was used to perform statistical analyses. The experiments were performed 3 times and the data were presented as mean±SD. Data from 2 groups were analyzed by unpaired t-test. Data from more than 2 groups were analyzed by one-way ANOVA. P value of <0.05 was considered to be statistically significant difference.
Results
Mutation-Derived Therapeutic Target Analysis in the DGIdb and Potential Role of Proteasome Inhibitors in ESCC Therapy
By combining single nucleotide variations (SNVs) and small insertion or deletion variations (indels) in the genomic sequencing data and the DGIdb database, a total of 307 therapeutic targeted genes in 425 out of 469 ESCC cases were identified. Of these druggable genes, 230 (75%) have Food and Drug Administration (FDA)-approved drug recommendations. All identified druggable genes with a mutation mutation frequency >1.5% are shown in Figure 1A. For the 50 mutated genes with a frequency >5%, only 5 genes had known drug-gene interactions. These genes included NOTCH1 (15.1%), LDL receptor related protein 1B (10.0%), PIK3CA (9.2%), CDKN2A (6.4%) and NOTCH3 (5.3%). All these 5 genes were previously identified to be significantly mutated genes in ESCC. 3 -7 In addition, only 5 out of 196 genes whose mutation frequency was 2.5-5% had known drug-gene interactions, including patched 1 (4.3%), erb-b2 receptor tyrosine kinase 4 (3.2%), NOTCH2 (2.8%), ATP binding cassette subfamily B member 1 (3.0%) and BRCA2 (2.6%); the first 3 genes were previously identified to be significantly mutated genes in ESCC (Figure 1A). 3 -7 However, most of the significantly and frequently mutated genes were potentially suppressor genes (such as CDKN2A and NOTCH1) and some of the recognized significantly mutated genes in ESCC had no targeted drugs (including ZNF750, ajuba LIM protein and FAT atypical cadherin 1) which possibly limited the potential as a therapeutic target. As a high consistency between different sources integrated by The DGldb suggested more marked clinical validity, the present study aimed to examine to what extent the 6 sources (DrugBank, PharmGKB, TEND, MCG, TALC and TTD) identified similar therapeutic target for given mutated genes; it was identified that only 4 genes [KDR, epidermal growth factor receptor (EGFR), BRAF and fms related tyrosine kinase 1 (FLT1)] had drug recommendations from all sources (Figure 1B). Although the mutation frequency was lower, KDR and EGFR harbored frequent and marked copy number amplification in ESCC. 17,18 The targeted therapy of EGFR in ESCC has attracted increasing attention; however, to the best our knowledge, an anti-KDR therapy has not been developed for ESCC so far.

Landscape of genomic SNVs, indels and therapeutic targets analyzed in the DGIdb. (A) Identified druggable genes in the DGIdb. The x-axis represents affected samples and the y-axis represents targetable genes recurrently mutated in at least 1.5%. The type of each mutation is shown for every sample, including the mutation frequency of specific genes (the right); Mutation subtypes are denoted by color. (B) Overlap between 6 sources integrated by The DGldb.
KEGG enrichment analyses of these druggable genes by DAVID identified some known significantly altered pathways in ESCC, including the receptor tyrosine kinase (RTK)-RAS, NOTCH, PI3K/AKT/mTOR, cell cycle and ERBB signaling pathways, which were enriched for most of the frequently and significantly mutated genes (Figure 2). In addition, the proteasome gene set was firstly identified to exhibit significant dysregulation in 8% of cases and 27 altered genes were potential targets for therapy, including proteasome subunit α 3, proteasome 26 S subunit, ATPase 2, proteasome 26 S subunit etc. Although these individual genes might not be identified to be significantly mutated genes because of the low mutation frequency, there are many variant family members with specific drugs targeting them. Interestingly, treatment with bortezomib (100 nM), a kind of 26 S proteasome inhibitor, resulted in a significant decrease in cell growth in 5 ESCC lines (Figure 3A). Furthermore, Eca109 cells were grown as tumor xenografts in BALB/c nude mice. After tumor establishment (5 x 5 mm), mice were administered 1.5 mg/kg bortezomib via intraperitoneal injection twice weekly. It was identified that bortezomib treatment significantly suppressed tumor growth and led to significant tumor regression (P < 0.05; Figure 3B and C). Consistently, it was noted that bortezomib treatment led to a significant reduction in Ki-67 expression in sections taken from the control and treated tumors (P < 0.01; Figure 3D). The results suggested a promising therapeutic merit of using a proteasome inhibitor in ESCC therapy.
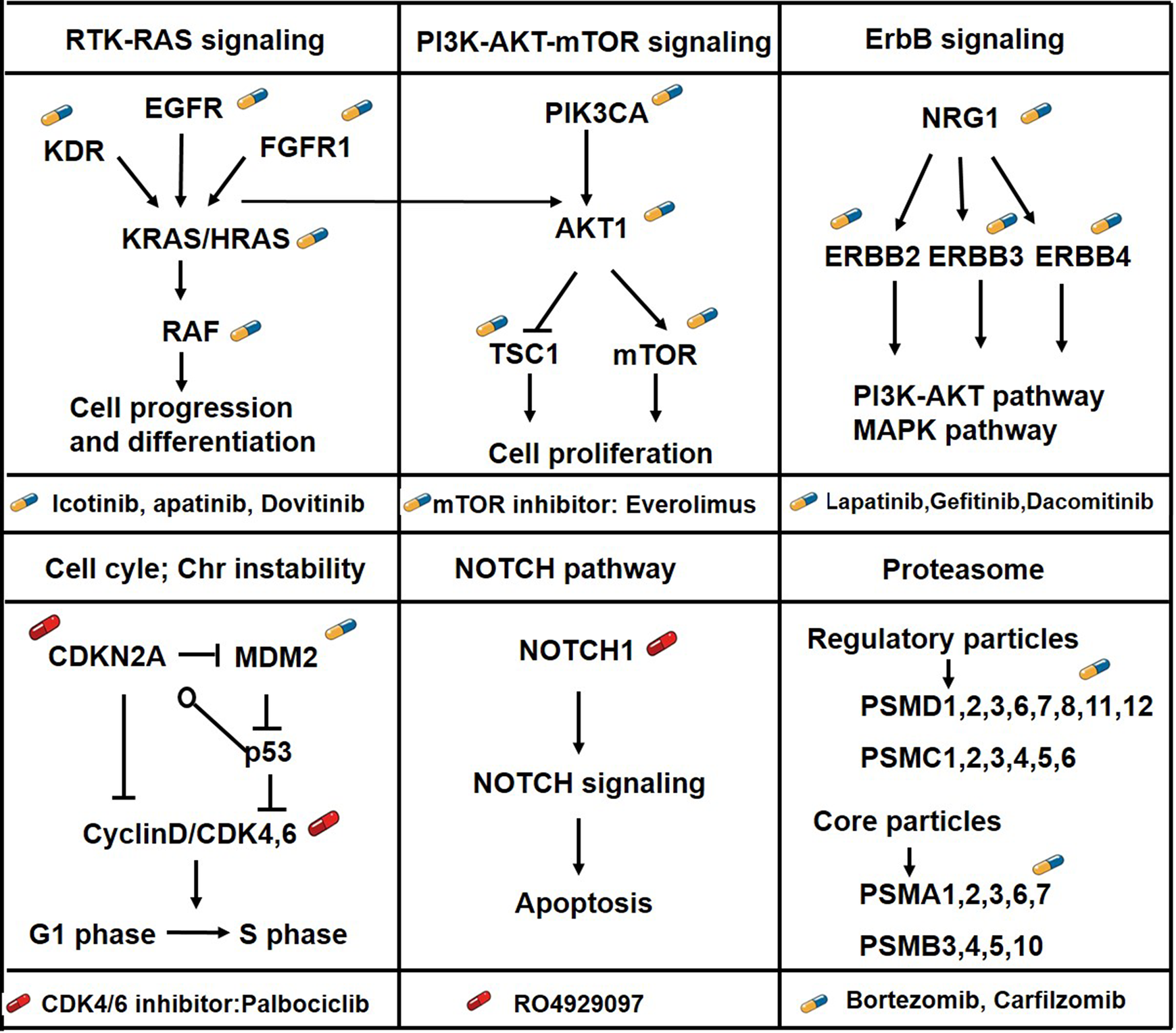
Targetable driving pathway analysis of druggable genes. The druggability is indicated by illustrations of pills; blue pills indicate the existence of FDA approved drugs and red pills indicate non-FDA approved drugs.

Proteasome inhibitor inhibits ESCC cell growth in vitro an in vivo. (A) MTT proliferation assay shows the effects of bortezomib treatment in 5 ESCC cell lines at 2 days (top) and 3 days (bottom). (B) Bortezomib induces regression of established human ESCC xenografts. The images represent the excised tumor appearance for each group. (C) Bortezomib significantly reduces tumor size and weight. The top graph shows tumor volume and the bottom graph shows tumor weight. (D) Representative immunohistochemical images show the expression level of Ki-67 of tumors from each group. The bottom panel shows the statistical histogram of the Ki-67 protein level. * P < 0.05, ** P < 0.01.
Potential Role of Targeting KDR in ESCC Cases With ZNF750 Variation
KDR, also known as VEGFR-2, was one of the 4 genes which had drug recommendations from all 6 sources integrated by The DGldb. In our previous study, the KDR gene was also found to be amplified in 4 out of 14 cases of a WGS set (28%; G score = 0.0257; Figure 4A). In the present study, it was identified that KDR might be a downstream target of ZNF750.

Identification of KDR as a downstream target gene of ZNF750. (A) Focal amplifications at the KDR locus (4q12) in 4 tumors of WGS are shown from The Integrative Genomics Viewer. (B) Detail of the heat map showing the cluster of seed-matched genes that were upregulated or downregulated by ZNF750 knockdown. The genes with fold changes >2 times are presented. (C) RT-qPCR was performed to monitor changes of KDR mRNA levels after ZNF750 was stably knocked down in KYSE140 (left) and KYSE150 (right) cells. (D) Protein expression analysis of KDR after ZNF750 was stably knocked down in KYSE140 (left) and KYSE150 (right) cells by western blot analysis. *P < 0.05, **P < 0.01.
ZNF750, a transcription factor which plays a key role in squamous epithelium late differentiation, was found to be frequently and significantly inactivated or mutated in ESCC. 3,5,19,20 Our previous experiments in vitro and in vivo demonstrated that ZNF750 inhibited tumor cell proliferation, growth, migration and invasion, acting as a tumor suppressor gene in ESCC. 3 In the process of studying the role of ZNF750 in ESCC, it was identified that ZNF750 knockdown resulted in a fold change >2 in the expression of 10 genes, including upregulated KDR using the human Cancer Pathway Finder RT 2 Profiler PCR array (Figure 4B). As shown in Figure 4C, the upregulation of KDR mRNA in stable ZNF750-knockdown ESCC cells compared with the control group was 5.87 and 4.23 fold change in KYSE140 and KYSE150 cells, respectively (P < 0.01). Moreover, western blot analysis further identified an upregulation of the KDR protein in stable ZNF750-knockdown ESCC cells (Figure 4D). These results suggested that KDR might be a downstream target of ZNF750. It is well known that there are many FDA-approved drugs targeting the KDR gene (including apatinib, cabozantinib, pazopanib and sorafenib) that have been applied to treat renal, gastric, colorectal and other cancers. 21 -24 However, KDR has seldom been previously implicated in ESCC therapy. To validate whether KDR is a downstream target gene of ZNF750, ZNF750 rescue experiments were performed by exogenously overexpressing ZNF750 and the effects on KDR were investigated. As presented in Figure 5A and B, rescue of wild-type ZNF750 in stable ZNF750-knockdown ESCC cells led to a marked restoration of KDR at the mRNA and protein levels (P < 0.01).

ZNF750 rescue restores KDR expression. (A) RT-qPCR results of KDR mRNA levels after ZNF750 rescue by its exogenous overexpression in stable ZNF750-knockdown KYSE150 (left) and KYSE140 (right) cells (Upper). The lower represents the relative mRNA expression of ZNF750 in 3 groups. ** P < 0.01. (B) Protein expression analysis of KDR after ZNF750 rescue in stable ZNF750-knockdown KYSE150 (left) and KYSE140 (right) cells by western blot analysis.
Our previous study demonstrated that ZNF750 knockdown could lead to increased cell proliferation and colony formation ability in ESCC. 3 Therefore, we performed KDR knockdown experiment in stable ZNF750-knockdown ESCC cells and the knockdown efficiency of KDR was validated using western blotting (Figure 6A). As exhibited in Figure 6B, knockdown of KDR significantly attenuated the promotion of ESCC cell growth caused by ZNF750 knockdown in both KYSE150 and KYSE140 cell lines. Consistently, knockdown of KDR in stable ZNF750-knockdown ESCC cells had a significantly decreased capacity for the formation of colonies compared with the control cells (Figure 6C). On the other hand, our previous study demonstrated that ZNF750 knockdown strongly promoted invasion and migration of ESCC cells. 3 Subsequently, it was investigated whether this promoting effect could be rescued by KDR knockdown. As hypothesized, the transwell assay showed that KDR knockdown could partly rescue the enhanced cell invasion induced by ZNF750 knockdown (Figure 7A). Consistently, the effect of accelerated migration caused by ZNF750 knockdown in ESCC cells was repressed by KDR knockdown, as determined by the wound healing assay (Figure 7B). All these data indicated that the phenotypes of ZNF750-knockdown ESCC cells could be reversed by KDR knockdown.

KDR knockdown inhibits growth of stable ZNF750-knockdown ESCC cells. (A) Knockdown of KDR in stable ZNF750-knockdown KYSE150 (left) and KYSE140 (right) cells was demonstrated by western blotting. (B) Growth assay shows inhibition of proliferation by KDR knockdown in stable ZNF750-knockdown KYSE150 (left) and KYSE140 (right) cells. (C) Colony formation assay with 3 experimental replicates revealed that knockdown of KDR significantly inhibited independent colony growth in stable ZNF750-knockdown KYSE150 (left) and KYSE140 (right) cells. * P < 0.05, ** P < 0.01.
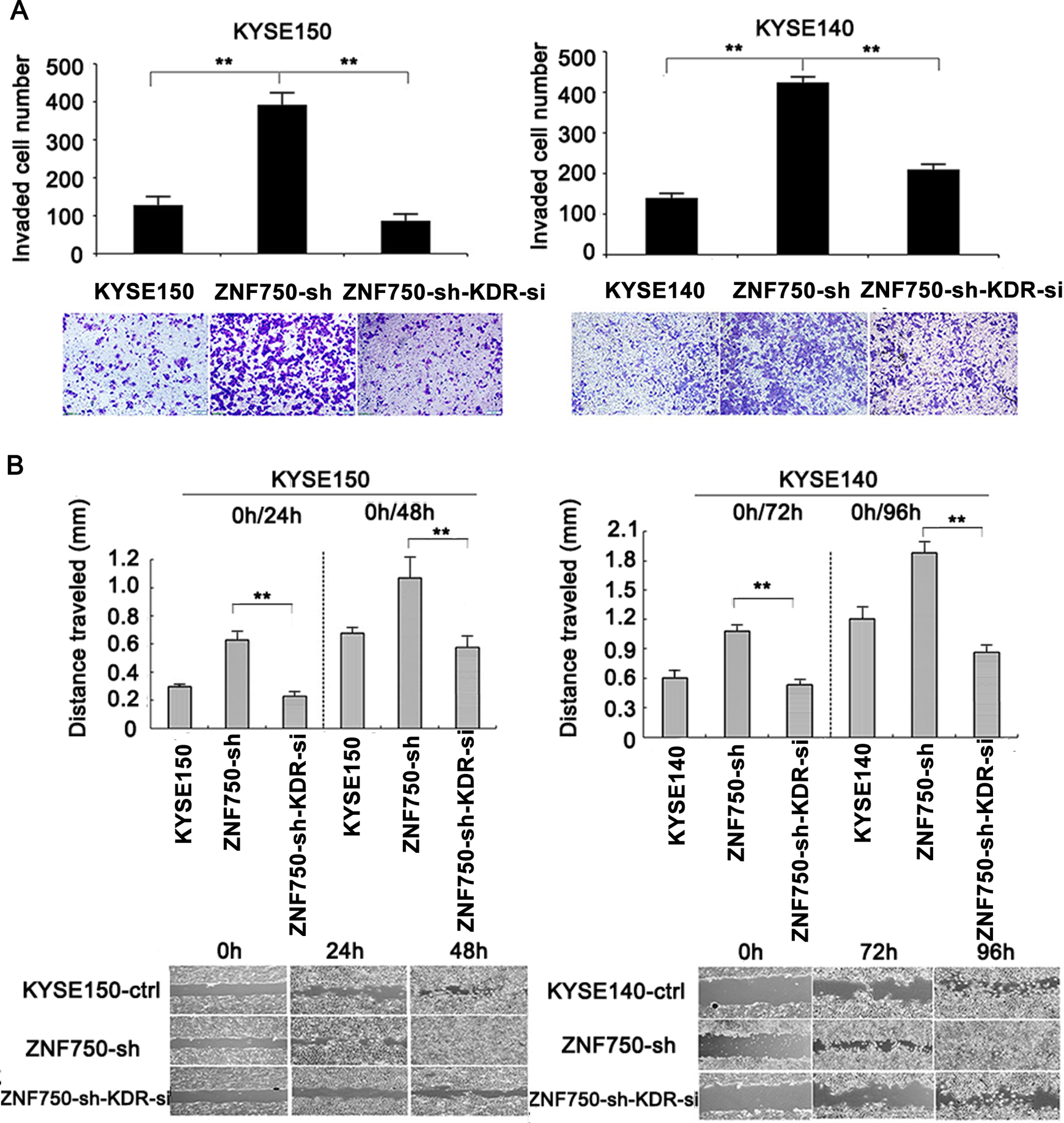
KDR-knockdown inhibits invasion and migration of stable ZNF750-knockdown ESCC cells. (A) Transwell chambers assay shows inhibition of invasion by KDR knockdown in stable ZNF750-knockdown KYSE150 (left) and KYSE140 (right) cells. (B) Scratch assay at specific time points revealed that knockdown of KDR significantly inhibited the migratory ability in stable ZNF750-knockdown KYSE150 (left) and KYSE140 (right) cells. * P < 0.05; ** P < 0.01.
In addition, apatinib, a kind of potent KDR inhibitor, was also used to further validate the role of targeting KDR in ESCC upon ZNF750 inhibition. We observed a suppressed viability on ESCC cells in a concentration-dependent manner. At concentrations of 15 and 25 μM, treatment with apatinib resulted in a profound inhibition of cell growth and colony formation, and the invasive effects were also significantly reduced (Figure 8A-C). Additionally, in a previous study, chromatin immunoprecipitation (ChIP)-sequencing of ZNF750 in squamous epithelium revealed that ZNF750 as a transcription factor bound the genes that it controls at a CCNNAGGC DNA motif. 25 Therefore, the ZNF750 binding sequence 2-3 kb upstream of the KDR transcriptional start site was identified and specific primers were synthetized. To determine whether the activity of KDR was regulated by ZNF750 by directly binding to its promoter, the ChIP assay was performed in KYSE150 cells overexpressing ZNF750 using an anti-HA tag antibody, followed by RT-qPCR, targeting KDR promoter regions. However, the assay was not able to demonstrate that ZNF750 directly bound to the KDR promoter (Figure 8D). Therefore, it was hypothesized that ZNF750 indirectly regulates KDR at the transcriptional level through other intermediate target molecules. The present results suggested that KDR may be an effective way to treat ESCC, especially for individuals with KDR amplification and ZNF750 inactive mutations.

Apatinib inhibits growth and invasion of ZNF750-knockdown ESCC cells. (A) Growth assay shows inhibition of proliferation by apatinib in stable ZNF750-knockdown KYSE140 (left) and KYSE180 (right) cells. (B) Colony formation assay revealed that apatinib treatment significantly inhibited independent colony growth in stable ZNF750-knockdown KYSE140 (left) and KYSE180 (right) cells. (C) Transwell chamber assay shows inhibition of invasion by apatinib treatment in stable ZNF750-knockdown KYSE140 (left) and KYSE180 (right) cells. Data are presented as the mean ± SD and each experiment was performed in triplicate *P < 0.05, **P < 0.01, ***P < 0.001. (D) Immunoblot analyses of HA protein in KYSE150 cells transfected with HA-tagged-ZNF750 (left); negative ChIP-PCR results of KDR (right).
Discussion
The occurrence and development of cancer is accompanied with abnormal genetic alterations causing the uncontrolled and progressive growth of somatic cells. Although most genetic alterations are passenger mutations that are not able to give cancer cells a selective growth advantage, an individual cell can gain a greater malignant phenotype if it acquires a sufficient series of genetic driving mutations. Therefore, targeting cancer-driving mutated genes and related pathways is very important for delaying cancer progression and metastasis. Recently, next-generation genome sequencing of cancer, including ESCC, improved the current understanding of molecular characteristics. 3 -7 Previously, the mutational landscape of 104 ESCC cases was determined, and a number of mutated genes and altered pathways associated with ESCC were identified. 3 However, the potential druggable targets and clinical validity of these genetic alterations have not been elucidated.
In this study, using the DGIdb, drug-gene interactions in genomic sequencing data were analyzed. However, most of the frequently and significantly mutated genes remain poorly targeted by existing drugs. This phenomenon limits these genes as clinical therapeutic targets. Additionally, the consistency of drug recommendations from different sources integrated in the DGIdb were compared and we found that there was very limited overlap between these databases, perhaps attributed to the different methodologies employed by different sources. It was observed that concordance was much greater when a large amount of strong evidence was available from clinical trials demonstrating that a specific drug acts directly on a given molecular variation, such as KDR and EGFR. In addition, KEGG enrichment analysis of all druggable genes identified some altered pathways in ESCC, including PI3K/AKT/mTOR, RTK-RAS, cell cycle, ERBB and NOTCH signaling. It has been reported that PI3K/AKT/mTOR signaling inhibitors (e.g. temsirolimus and everolimus) could inhibit growth of ESCC in vitro and in vivo, indicating that this pathway may provide a novel target for the treatment of ESCC. 26 -28 The RTK-RAS signaling was also found to be enriched by druggable genes. A total of 2 upstream members, EGFR and ERBB2, have been shown to harbor copy number gains and are overexpressed in ESCC. 29,30 Two categories of anti-HER-family-targeting therapies have been in clinical development and some EGFR TKIs, such as erlotinib and gefitinib, have shown better curative effects in ESCC. 31,32 In the present study, the proteasome was also identified as a potential therapeutic target in ESCC. Proteasome inhibitors have been applied to the therapy for a variety of tumors, including acute myeloid leukemia, multiple myeloma and colorectal cancer, but not ESCC. 33,34 The present results demonstrated that proteasome inhibitors could markedly inhibit the proliferation of ESCC in vivo and in vitro, and provided novel insight for targeted therapy of ESCC. This result is consistent with the previous results that inhibition of the proteasome can induce autophagy in human ESCC cells and also increase cell death. 35
Among numerous targetable genes, KDR gene was simultaneously identified by different sources integrated by The DGIdb. KDR, also known as VEGFR-2, plays critical roles in tumor angiogenesis by mediating the interaction with VEGF and directly impacts cell proliferation through regulating autocrine/paracrine processes. 36 -38 KDR has been reported to be upregulated in various cancer types and acts as an oncogene, with 100% positive expression in ESCC. 12 -14,39 Currently, anti-KDR agents (such as IMC-1C11 and elpamotide) have been used in clinic therapy for various cancer. In advanced-stage gastroesophageal cancer, KDR is a clinically validated molecular target. 40 In ESCC, Xu et al indicated that the usage of species-specific neutralizing antibodies for KDR could suppress ESCC tumor growth, metastasis and angiogenesis in preclinical models, providing strong evidence of the importance of tumor micro-environments in cancer progression. 41 In the present study, it was further determined that KDR might be a downstream target of ZNF750.
ZNF750 has been empirically proved to harbor frequently inactivating mutations in different types of human squamous cell carcinomas by previous independent studies. 3,6 ZNF750 is a nuclear protein that acts as a transcription factor playing an important role in maintaining and controlling squamous epithelium homeostasis by inhibiting progenitor genes while inducing differentiation genes. 42 In our previous study, mutations of ZNF750 were identified in 6% of ESCC tumors and ZNF750 deletions were observed in 21% of ESCC tumors. The inactivating variation of ZNF750 might confer growth advantage on ESCC cells and were under positive selection in the evolution process of ESCC. 3 Previous functional studies also showed that wild-type ZNF750 strongly inhibited ESCC cell proliferation, migration and invasion. 3,6 Although these results were consistent with other previous results, the downstream targets of ZNF750 in ESCC were not fully investigated. Hazawa et al found that long non-coding RNA, TINCR ubiquitin domain containing and laminin subunit γ 2, mediated the growth inhibition and migration suppression of ZNF750 in ESCC, respectively. 19 In the present study, ZNF750 knockdown led to the marked upregulation of KDR expression and rescue of wild-type ZNF750 led to marked restoration of KDR in ESCC cell lines. Additionally, the ZNF750-knockdown phenotype was reversed by depletion of KDR. Although ZNF750 was not able to bind to the promoter region of KDR directly, the present results suggested that the tumor suppressor role of ZNF750 gene in ESCC might be partly mediated by KDR indirectly. The phenomenon that KDR could be regulated by ZNF750 in ESCC suggested that targeting KDR overexpression might be an effective way to treat ESCC, especially for KDR variant cases and in individuals with ZNF750 mutations or deletions.
It should be pointed out that the patients evaluated in this study are all from Asian countries, in which ESCC is the dominant histological type. In western countries with high incidence of esophageal adenocarcinoma, ESCC is relatively rare. Because of this, almost all the ESCC samples of whole genome sequencing were from the Asian countries such as China, Iran, India, Japan. There are few reports of whole genomic sequencing on the samples from western countries. Although whether there is a difference between the etiology of ESCC in Asian and Western countries is unknown, our results may provide potential therapeutic targets for ESCC in both Asian and Western countries. Of course, further investigation are required for these therapeutic interpretation for western populations.
In summary, genomic sequencing data from the DGIdb were analyzed, and the possible application value of proteasome inhibitors and targeting KDR in the treatment of ESCC was assessed. Due to the lack of in-depth research and clinical experiments, further investigation is required for the therapeutic interpretation of these target genes.
Footnotes
Abbreviations
Authors’ Note
The use of experimental animals was approved by the Ethics Committee of Shanxi Medical University (Reference # 2017LL037) and strictly followed the guideline for tumor induction in mice and rats.
Acknowledgments
The authors would like to thank Dr Bin Song(The First Hospital, Shanxi Medical University, Taiyuan, Shanxi) for their help and provide apatinib in our experiments.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the National Natural Science Foundation of China (81672768 to X.C., 81773150 to L.Z., 81602175 to H.L.), the Program for the Outstanding Innovative Teams of Higher Learning Institutions of Shanxi (OIT 2017 to L.Z.), the Doctoral Start up Research Fund of Shanxi Medical University (03201508 to L.Z.), the Science and Technology Innovation Fund of Shanxi Medical University (01201310 to L.Z.).