
Abstract
The McMurdo Dry Valleys may harbor diverse surface microbial communities, yet little is known about subsurface microorganisms in permafrost and their potential for paleoecological reconstruction. Here, we present microbial diversity and paleoecology from lower Wright Valley (7000- to 25,000-year-old) and Pearse Valley (>180,000-year-old) permafrost habitats in the McMurdo Dry Valleys. Using a new decontamination protocol, low-biomass extraction approaches, and 16S ribosomal RNA gene amplification sequencing, we assessed microbial community structure and diversity. The difference between surface and subsurface microbial communities at both lower Wright and Pearse valleys suggests the environmental conditions were different at the time of colonization. Microbial taxa identified in subsurface permafrost but not in the surface soil in both valleys indicate an ancient and isolated microbial community. In contrast, communities were not resolved at a high-elevation site in the stable upland zone, the Friis Hills (>6 Ma). The inability to identify DNA using amplicon sequencing in the Friis Hills is consistent with previous efforts to analyze high-elevation soils and permafrost, which suggests that microbial habitability is severely restricted in persistent cold, arid habitats. Therefore, utilizing other approaches may be necessary to analyze surface and subsurface permafrost on Earth, and perhaps Mars, where low-abundance microbial populations may be present.
Introduction
Permafrost (perennially frozen) archives extend beyond ice core records and may hold valuable information regarding terrestrial climate change (Gilichinsky et al., 1992). Therefore, the information preserved within permafrost may be useful to constrain boundary conditions for ice sheet models to determine surface melt and resolve uncertainties regarding ice sheet contributions to sea-level rise (DeConto and Pollard, 2016; Lowry et al., 2021). These permafrost environments contain frozen reservoirs of ice, greenhouse gases, ancient bacteria, and viruses (Adriaenssens et al., 2017; Gilichinsky et al., 2007; Ruggiero et al., 2023). Future thawing of these environments due to increasing atmospheric temperatures could increase microbial activity and release previously frozen gases and nutrients, which could lead to unprecedented changes in hydrological and biogeochemical cycles.
Frozen strata (cryostratigraphy) record the last time the landscape was unfrozen, and DNA from microbes within these frozen strata may correspond to the time of their formation (since freezing). While microbial communities may have adapted to live in permafrost and represent postdepositional metabolism (Mackelprang et al., 2017), they may have colonized an environment that is different from the present surface. Slow reproduction, metabolism, and evolution of microorganisms at low temperatures allow observation of biological processes on geological timescales (Gilichinsky et al., 1992, 2007). If a robust understanding of permafrost age, stability, and evolution through time can be obtained, then the relationship between microbial populations and their permafrost habitat can be established. Therefore, preservation of microbial characteristics, both in terms of abundance and diversity, can be regarded as a biomarker of climatic and paleoecological conditions during the time of colonization, erosion, and sediment deposition (Gilichinsky et al., 1992; Wilson et al., 1996). Once established, the occurrence of microbial populations in a habitat that forms part of the frozen strata can contain paleoecological and chronological information and can be used to correlate with other stratigraphic records (e.g., Gilichinsky et al., 1992; Wilson et al., 1996).
The McMurdo Dry Valleys are a hyperarid, cold, polar desert and can be subdivided into three geographic zones (coastal thaw, inland mixed, and stable upland), which are defined by their microclimatic parameters of atmospheric temperature, soil moisture, and relative humidity (Fig. 1; Marchant and Denton, 1996; Marchant and Head, 2007). High elevations (>1000 meters above sea level [masl]) are defined as the stable upland zone in the McMurdo Dry Valleys (Marchant and Head, 2007). Previous studies of permafrost in the stable upland zone have identified microorganisms from University Valley that utilize phospholipid fatty acid biomass (Tamppari et al., 2012), and cultured microbial populations have been isolated from University Valley (Goordial et al., 2016) and Beacon Valley and Mount Feather that may be >5 Ma (Gilichinsky et al., 2007). Due to low temperatures (<0°C), thin films of unfrozen brine, and the absence of ultraviolet (UV) radiation penetrating >5 mm below the surface (Nienow et al., 1988), Antarctic permafrost in the stable upland zone is suitably stable and cold to preserve ancient DNA (Gilichinsky et al., 2007; Tamppari et al., 2012) or may reflect active DNA repair as a long-term survival mechanism (Johnson et al., 2007). These field observations are supported by laboratory incubation experiments that have shown metabolism continues down to −15°C (Rivkina et al., 2000) and have speculated possible low rates of metabolism at temperatures as low as −25°C (Price and Sowers, 2004). However, Goordial et al. (2016) could not detect microbial activity at in situ temperatures in high-elevation ice-cemented permafrost in University Valley, compared with activity in low-elevation Dry Valley permafrost soils. In the Shackleton Glacier region of the Transantarctic Mountains, cultivation-dependent and cultivation-independent methods were also unable to detect viable microbial life at high elevation, which suggests that life may be at the margins of habitability in the high-elevation soils at Roberts Massif and Shroeder Hill (Dragone et al., 2021, 2022). These studies suggest a potential limit of microbial habitability, where microbial life is severely restricted in high-elevation soils with persistent cold and arid conditions. However, the authors of these studies acknowledged that their protocols may not have been optimized to detect microbial DNA from extremely low-biomass permafrost soils.
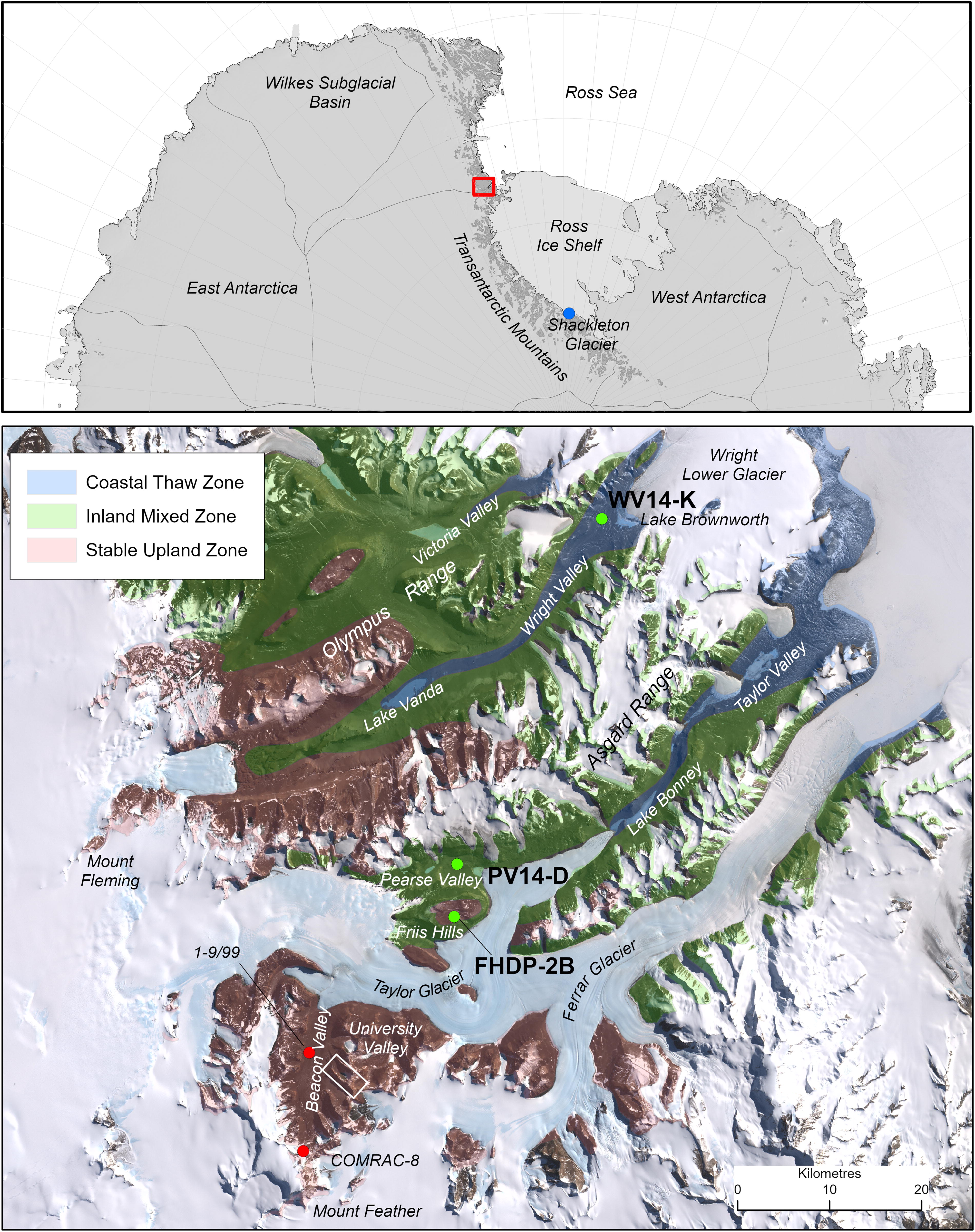
McMurdo Dry Valleys study area and associated sites discussed in the text. Green circles show the locations of the lower Wright Valley, Pearse Valley, and Friis Hills sites where permafrost cores in this study were recovered. Red circles show the locations of permafrost cores 1–9/99 from Beacon Valley and COMRAC-8 from Mount Feather (Gilichinsky et al., 2007). White box shows the location of University Valley. The three microclimatic zones are the stable upland zone (brown), inland mixed zone (green), and coastal thaw zone (blue). Modified from Marchant and Head (2007); and Salvatore and Levy (2021).
Though surface soils in Antarctica have a wide diversity of microbial communities (Cary et al., 2010), knowledge about diversity in subsurface permafrost is largely restricted to culture-based studies (e.g., Gilichinsky et al., 2007; Goordial et al., 2016). To date, few studies have successfully used a culture-independent approach to examine diversity in high-elevation Dry Valleys permafrost (Goordial et al., 2016, 2017). A limitation of culture-based studies is that it is challenging to recreate the conditions required to promote growth of all taxa present, so these culture-based studies do not accurately characterize the true abundance and diversity of bacteria in the McMurdo Dry Valleys (Smith et al., 2006). In fact, many phylotypes identified by 16S ribosomal RNA (rRNA) gene analysis have not been identified in culture-based community studies (Smith et al., 2006). Therefore, we are compelled to ask two questions. (1) Is there a limit to microbial life, or are we limited by our approach to identify these low-biomass taxa? (2) Can we develop an ultra-low-biomass DNA extraction method to characterize microbial communities from permafrost using culture-independent methods in the stable upland zone?
Three problems that complicate current methods for Antarctic soil studies and analysis of permafrost in hyperarid, polar environments are contamination, low-biomass, and difficulty differentiating between DNA extracted from external contaminants, extant permafrost microorganisms, and DNA preserved in the environment (e.g., Dragone et al., 2021; Goordial et al., 2016; Mackelprang et al., 2017). In this study, we address the first challenge, contamination in permafrost samples, by presenting a novel decontamination protocol. We demonstrate the efficacy of this approach and report both the microbial diversity and community structure of permafrost and surface soil from two locations that differ in geological history. We present 16S rRNA gene amplicon analysis to study microbial populations in permafrost cores from lower Wright Valley (WV; 7–25 ka) and Pearse Valley (PV; >180 ka) in the McMurdo Dry Valleys. At Friis Hills (>6 Ma), in the stable upland zone, the method was not able to detect DNA using amplicon sequencing. Using this approach, we demonstrate that external contaminants can be controlled. Despite being unable to differentiate between the physiological states of the DNA, active or dormant communities reflect adaptations to life in permafrost (since freezing) or the time the enclosing strata became frozen, but not the current surface soil habitat (Mackelprang et al., 2017). Results from ultra-low-biomass communities, such as those found in the McMurdo Dry Valleys, are of special interest in understanding microbial diversity in harsh, hyperarid, polar climates. As such, they also represent the best analog on Earth to test and better understand the potential for habitability of martian permafrost.
Sample sites
The lower WV core (WV14-K) was collected at 326 masl (77.4252°S, 162.6664°E), ∼2 km west of Wright Lower Glacier (Fig. 1). Radiocarbon dates of lacustrine algae give an age of ∼7–25 ka for shallow permafrost horizons (Hall et al., 2001). The PV core (PV14-D) was located on an elevated bench that extends along the northern side of the valley floor at 450 masl (77.7062°S, 161.5467°E), ∼3 km northwest of the present position of the Taylor Glacier lobe. In situ cosmogenic nuclide depth profiles give an age of >180 ka for permafrost >2.09 m depth (Anderson et al., 2023). Sediments from lower WV and PV cores comprise ice-cemented permafrost, with grains of sand and pebbles forming the matrix, and the pore spaces filled with ice (Anderson et al., 2023). The geologic setting and characteristics of the lower WV and PV permafrost cores were described by Anderson et al. (2023). The Friis Hills core (FHDP-2B) was located at 1296 masl (77.754715°S, 161.455706°E). The permafrost sediments comprise mid-Miocene (∼14 Ma) interbedded diamict and fluvio-lacustrine sediments (Chorley et al., 2022). While the sediments at Friis Hills are dated to the mid-Miocene (Chorley et al., 2022), warm intervals may have caused surface melt in the late Miocene, which suggests the permafrost has been frozen for >6 Ma (Dickinson et al., 2012; Verret et al., 2023). The modern climate characteristics of these sites are summarized in Table 1.
Modern Climate Characteristics of Three McMurdo Dry Valley Sites
Modern Climate Characteristics of Three McMurdo Dry Valley Sites
Hall et al. (2001).
Obryk et al. (2020).
Fountain et al. (2010).
Anderson et al. (2023).
McKay unpublished data.
Verret et al. (2023).
McKay et al. (1998).
masl = meters above sea level.
Surface samples (SSs; 2–7 cm depth) were collected after the armoured surface layer of desert pavement (∼2 cm depth) was removed using an ethanol-sterilized trowel and metal spatula. Samples were put into a 50 mL Falcon tube prior to drilling at each site. During the 2014/2015 austral field season, ice-cemented permafrost cores were aseptically recovered from lower WV and PV with the use of a gasoline powered dry rotary drilling technique (Anderson et al., 2023) (Fig. 1). During the 2016/2017 austral field season, a permafrost core was recovered from the Friis Hills with the use of a cold weather modified, Webster Drilling HPP-150 drill rig and chilled compressed air as drilling fluid (Chorley et al., 2022). Each permafrost core was extracted from the core barrel in lengths between 0.1 and 0.5 m and split using an ethanol-sterilized chisel into ∼0.1 m sections where it maintained structure. Permafrost core samples had a ∼7 cm diameter and ∼9 cm height and were collected in a combination of sterile Whirl-Pak bags and bleach-sterilized polyvinyl chloride core liners. Gravimetric water content (in wt%) was 11–18% for ice-cemented permafrost from lower WV (WV14-I) and 6–15% for PV (PV14-A). The gravimetric water content was 5–40% for Friis Hills (FHDP-2C) (Verret et al., 2021). Subsurface ice-cemented permafrost samples for microbial analyses were collected from depths beneath the surface of 1.53–1.63 m (lower WV), 2.83–2.94 m (PV), and 0.185–0.25, 0.25–0.32 m (Friis Hills), respectively. Subsurface samples from Friis Hills (FHDP-2B) were collected directly below the dry permafrost (defined as having negligible ice content), targeting ice-cemented organic-rich interstratified mud-sandstone units (Chorley et al., 2022). The samples were kept frozen at −80°C in the laboratory until processing and analysis.
Decontamination protocol
A stringent decontamination protocol was specifically designed for this study to ensure that amplified microbial taxa were sourced only from the permafrost core and not from external contamination prior to or during DNA extraction (Fig. 2). To achieve this, the outer layer of permafrost was contaminated with ∼10 mL of viable Escherichia coli carrying a quantitative PCR (qPCR) reportable pGEM plasmid (pGEM-3Z Vector). The pGEM contaminant was applied over the outer surface of the core using a Pasteur pipette. The contaminant consisted of a total of 1 mL of culture medium, which consisted of 500 µL of high-density (2 × 109 cells/mL) E. coli pGEM culture and 500 µL of 50% glycerol that was diluted in 10 mL of phosphate-buffered saline. Each core was then individually and rigorously decontaminated via a series of six consecutive wash baths to remove the pGEM contamination. In a step-wise approach, each core was fully submerged and rinsed in each bath for ∼30 s, starting in a bath of 1:3 bleach and then followed by a 1:8 bleach bath, sterile water bath, second sterile water bath, sodium thiosulfate bath (to neutralize any residual bleach), and a final sterile water bath. By the final wash step, between ∼1 and 2 cm of the outer core had been physically removed. The initial frozen weight of each permafrost core was >200 g. The exact weight and volume could not be obtained without risking thaw and contamination of each sample. Control samples were collected in the biosafety cabinet from the second sterile water wash and the final sterile water wash. Each control was used to detect if either the pGEM contaminant or any external contaminants were introduced during the procedure. After the decontamination protocol, each permafrost core was placed in an autoclaved and UV-sterilized melt jar that contained a clean Teflon magnetic stir bar for sample fractionation. Approximately 200 mL of sterile water was then poured into the jar to submerge the sediments. Following closure, the melt jar was placed in an ultrasonic bath for 3 min and stirred for 30 min using the magnetic stir bar. After allowing the sediments to settle for 2–4 h, water and fine-grained surface sediments were removed from the melt jar using a peristaltic pump and spinal tap needle. The collected core water and wash step control samples were then spun down and stored at −80°C until DNA extractions were performed (see Supplementary Data S1 for the detailed decontamination protocol and DNA extraction procedures).
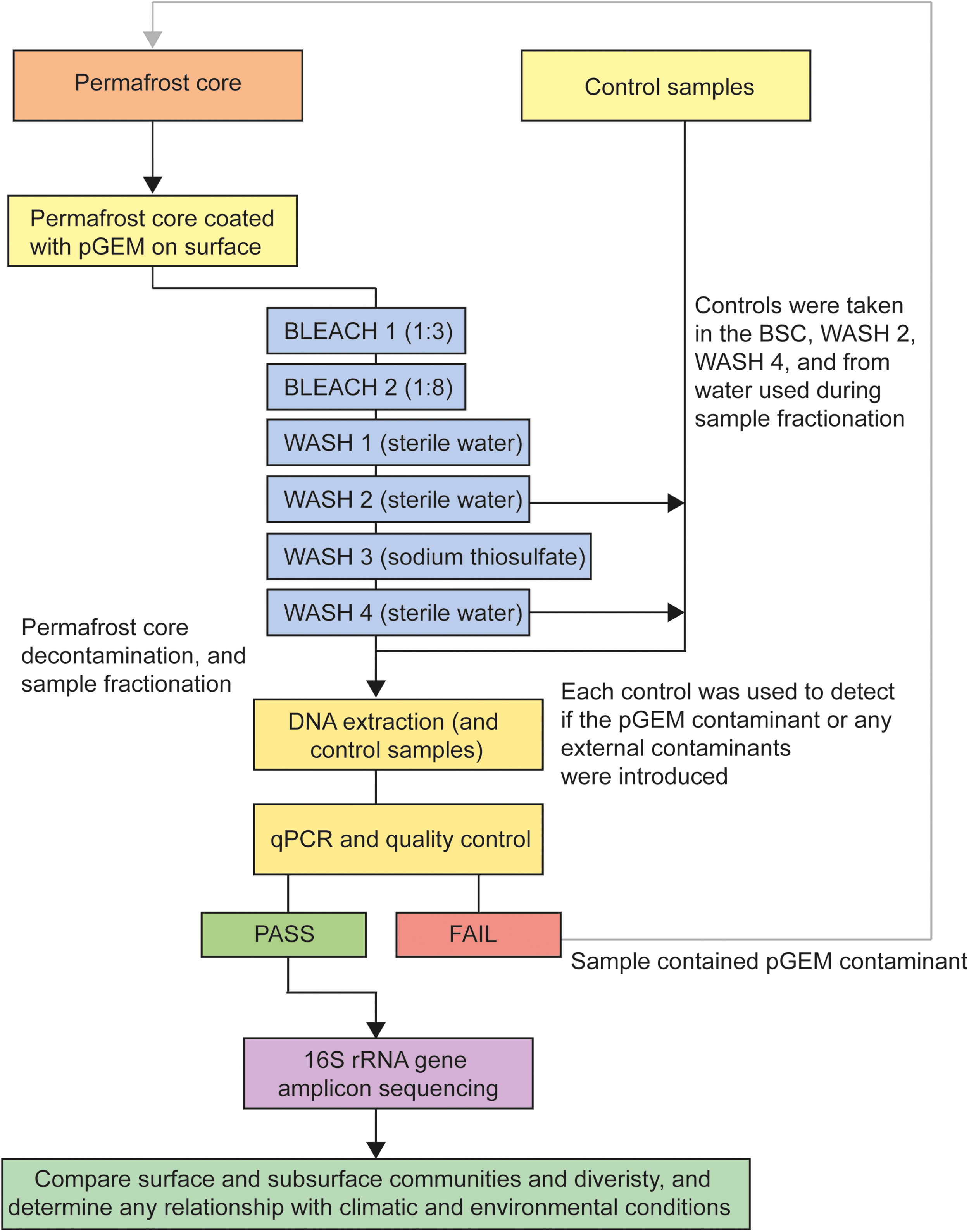
Experimental design for permafrost core decontamination, DNA extraction, and microbial community analysis.
Total DNA was extracted from SS and subsurface sample from lower WV, PV, and Friis Hills (FHDP). For lower WV and PV SSs, total DNA was extracted from ∼1 g of soil, using a modified CTAB (cetyl-trimethyl-ammonium-bromide) bead-beating method (Coyne et al., 2001). For Friis Hills sample FHDP SS-1, DNA was extracted from ∼5 g of soil, washed with Tris-HCl/EDTA mixture, and spun; then the supernatant was removed, and DNA was extracted from ∼1 mL using the modified CTAB bead-beating method (Coyne et al., 2001). After the permafrost core decontamination protocol, subsurface permafrost samples and decontamination wash controls were defrosted on ice to room temperature and transferred into 1.5 mL screw-capped conical bottomed polypropylene tubes that contained 0.5 g each of 0.1 and 2.5 mm UV-sterilized silica–zirconia beads. Total DNA was extracted from ∼1–2 mL of each permafrost extract using the CTAB bead-beating method (Coyne et al., 2001). Samples >1 mL were pooled after the addition of 500 µL chloroform:isoamyl alcohol (24:1), vortex, rocking, centrifuge, and removal of the upper aqueous layer into a new 1.5 mL sterile microfuge tube. All extracted DNA samples and wash bath controls were quantified with Qubit Fluorometer double-stranded (dsDNA) high-sensitivity (HS) assay kit (Thermo Fisher Scientific) (Table 2). QPCR targeted to the M13F/pGEMr region of the pGEM plasmid were quantified in the sample and wash baths 2 and 4. The following qPCR conditions for the detection of pGEM were used: 95°C for 2 min, followed by 55 cycles at 95°C for 10 s, and 60°C for 40 s. Samples with no residual pGEM were then quality controlled using qPCR detection of the 16S rRNA gene using the Earth Microbiome Project (EMP) primer set 515F and 926R (Table 2). The following qPCR conditions for the detection of the 16S rRNA gene were used: 94°C for 3 min, followed by 30 cycles at 94°C for 45 s, 50°C for 1 min, 72°C for 1.5 min, and a final extension step at 72°C for 10 min. Only samples that met the following conditions proceeded to 16S rRNA sequencing: (1) did not amplify pGEM after 50 qPCR cycles, (2) did amplify a 16S rRNA signal within 30 qPCR cycles, (3) the corresponding final wash bath 4 and DNA extraction blanks did not amplify pGEM, and (4) the DNA extraction blanks did not amplify 16S rRNA.
DNA Extraction Quality Control, 16S Ribosomal RNA Sequencing, and Sequenced Reads for All Samples and Controls
DNA Extraction Quality Control, 16S Ribosomal RNA Sequencing, and Sequenced Reads for All Samples and Controls
Detection limit for Qubit high sensitivity is 0.005 ng/µL.
BME = beta-mercaptoethanol; CTAB = cetyl-trimethyl-ammonium-bromide; QC = quality control; qPCR = quantitative PCR; rRNA = ribosomal RNA.
The microbial communities were amplified in permafrost, surface soil, and extraction blank controls through targeting the V4 region of the 16S rRNA gene using the EMP primer set 515F and 926R following the method of Monteiro et al. (2021). They were sequenced using an Ion Torrent 530 chip chemistry at Penn State University. Prior to amplification, the qPCR master mix and each fusion primer were confirmed to be contaminant free via running a DNA negative qPCR. The sequencing PCR was prepared in a laminar flow hood while the operator wore a contamination suit, face mask, and eye protection. Samples were prepared from the same master mix in triplicate, with the extraction blanks prepared first, followed by samples in increasing order from the lowest to the highest extracted DNA concentration. For each reaction, a negative control was amplified and confirmed to be contaminant free via gel electrophoresis (Supplementary Fig. S1). To determine if any potential contamination occurred during the extraction or amplification process, after amplification, each amplified extraction control sample was tested for contamination via qPCR and high-resolution melt. Samples and extraction blank controls were each sequenced to an average depth of ∼400,000 reads (Table 2). The raw DNA sequences presented in this study can be found under the BioProject accession number PRJNA1152789.
The presence of either a forward or reverse V4 16S rRNA PCR primer was confirmed and trimmed from each read with Cutadapt v2.3 (Martin, 2011). Amplicon sequence variants (ASVs) were generated with reads <230 bp, quality score <2, and expected error >2 removed using DADA2 v1.14.1 (Callahan et al., 2016) in R v 4.1.0 (Team RC, 2021). Taxonomy was assigned to ASVs with DECIPHER v2.22.0 (Wright, 2016) using the SILVA v138 database (Quast et al., 2013). Sequences were aligned using multiple alignment using fast Fourier transform v7 (Katoh et al., 2002). Data analysis was conducted in R version 4.1.2 with the following packages: kableExtra v1.3.4; eulerr v6.1.1; breakaway v4.8.4; tidyr v1.2.1; tidyverse v1.3.2; ggplot2 v3.4.0; and phyloseq v1.38.0. Data were processed on the New Zealand eScience Platform.
Results
16S rRNA community analysis
Fluorescence-based quantifications (Qubit and qPCR targeted to the 16S rRNA gene) were used to confirm the presence of DNA in all samples prior to sequencing (Table 2). Ion Torrent sequencing produced between 1172 and 884,479 raw sequence reads. The depth of the Ion Torrent 530 sequencing chemistry adequately resolved the presence of low-level contaminants (Supplementary Fig. S2) within control extraction blank samples using manual curation against a list of common reagent and human contaminants (Salter et al., 2014) and using the statistical approach against the control samples within the R package Decontam (v1.14.0) (see Supplementary Data S2). A single prevalent ASV (ASV_3) associated with the genus Pseudomonas was identified as a contaminant within the extraction controls. These Pseudomonas read counts (extraction blank sample number = 5; sequenced read count average = 12,925 ± 1966) are representative of ∼2.7% of the average total read count for the true samples (sample number = 4; 475,879 ± 63,601). In total, 52 ASVs were flagged as potential contaminants and were identified and removed from the dataset (highlighted red in Supplementary Data S2). This made up <1% of the total reads. Contaminants identified in the controls that were not listed as common contaminants included Alishewanella, Cutibacterium, Halomonas, and Lautropia. After quality control and contaminant removal, 363,743–459,552 reads remained in SS and subsurface sample collected from lower WV and PV. In the Friis Hills samples, only three to nine reads associated with the bacterial phyla Actinobacteriota, Bacteroidota, Chloroflexi, Desulfobacterota, Firmicutes, and Proteobacteria were retained after quality control (Table 2 and Supplementary Data S2).
Microbial diversity at lower Wright Valley and Pearse Valley
Bacteria dominated the microbial communities in both surface soils and subsurface permafrost at lower WV and PV and were predominantly Acidobacteriota, Actinobacteriota, Bacteroidota, and Proteobacteria. Archaeal taxa comprised <1% of the total relative abundance and were predominantly members of the ammonia oxidizer phylum Crenarchaeota (genus Candidatus Nitrosocosmicus). Thermoplasmatota and Halobacterota were rare and restricted to subsurface permafrost.
The most abundant bacterial phyla in the lower WV surface soil sample (WVSS1) were Bacteroidota (46.4%), Actinobacteriota (29.1%), Proteobacteria (15.2%), and Deinococcota (5.8%) (Fig. 3). Within Bacteroidota, the most abundant genus was Ferruginibacter. Within Actinobacteriota, the most abundant genus was Rubrobacter, a radiotolerant extremophile. Within Deinococcota, the most abundant genus was Truepera, which has resistance mechanisms that can withstand ionizing (UV) radiation.
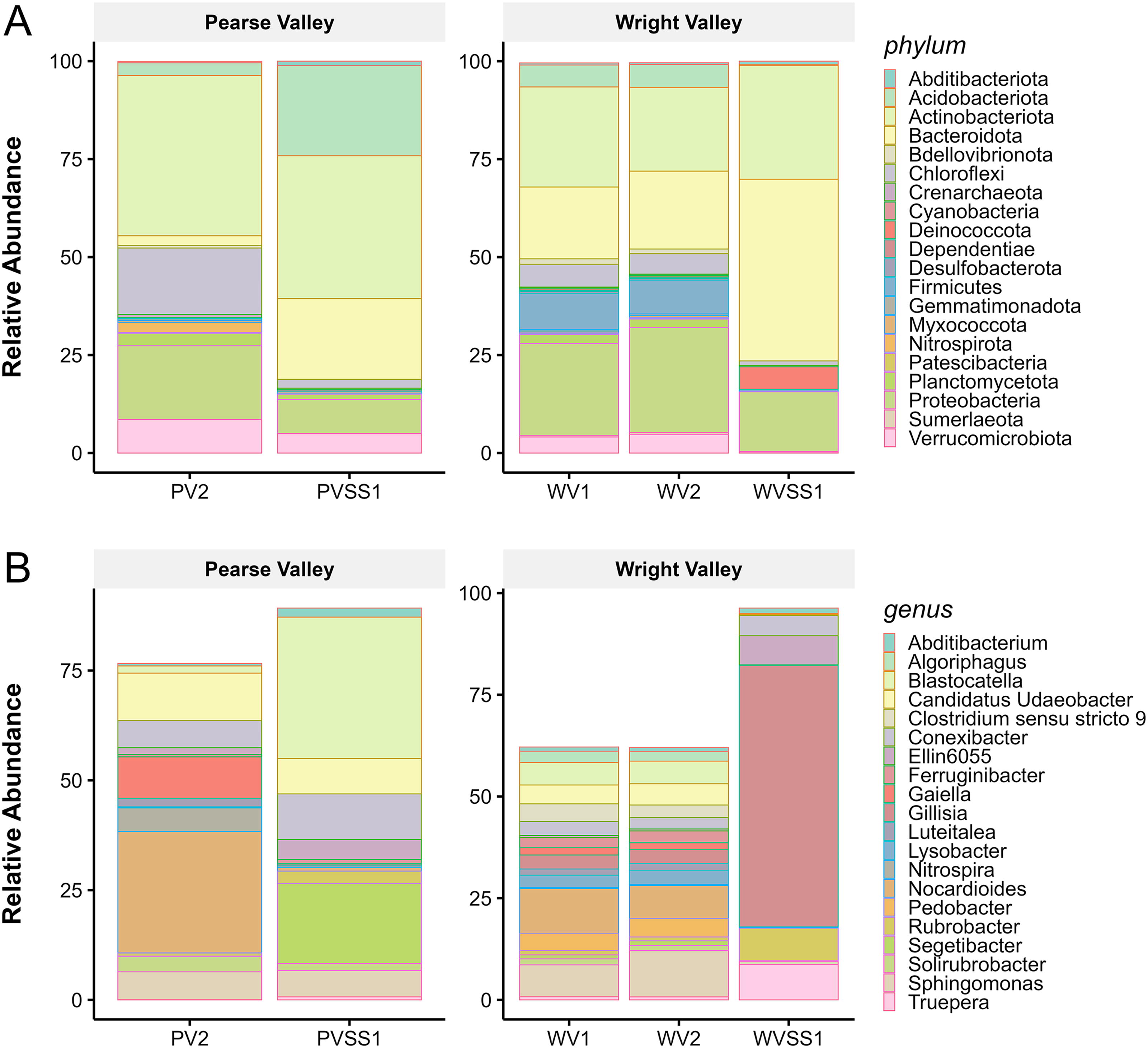
Relative abundance of the 20 most dominant bacterial
The most abundant phyla in the PV surface soil sample (PVSS1) were Actinobacteriota (36.5%), Acidobacteriota (23%), Bacteroidota (20.6%), and Proteobacteria (8.7%) (Fig. 3). Within Acidobacteriota, the most abundant genus was Blastocatella. Within Bacteroidota, the most abundant genus was Segitobacter, which is associated with dry soil conditions (Monteiro et al., 2021). Within Actinobacteriota, the most abundant genus was Conexibacter. Within Verrucomicrobiota, the most abundant genus was Candidatus Udaeobacter. Monteiro et al. (2021) found order Blastocatellales exclusively associated with dry and transition zone soils in the McMurdo Dry Valleys.
The subsurface ice-cemented permafrost communities demonstrated higher diversity than the surface soil samples at both lower WV and PV (Fig. 3) The most abundant phyla for the lower WV subsurface permafrost sample (WV1) were Actinobacteriota (25.6%), Proteobacteria (23.6%), Bacteroidota (18.3%), Firmicutes (9.3%), Chloroflexi (5.8%), and Acidobacteriota (5.6%). For the lower Wright Valley subsurface, the most abundant ASVs were within the genera Nocardioides, Sphingomonas, Pedobacter, Candidatus Udaeobacter, Blastocatella, and Clostridium sensu stricto 9. Sphingomonas are associated with dry soil conditions in the McMurdo Dry Valleys (Monteiro et al., 2021). The most abundant ASVs were within families Chthoniobacteraceae, Comamonadaceae, Nocardioidaceae, and Xanthomonadaceae. Clostridium sensu stricto 9 (Firmicutes), Pedobacter (Bacteroidota), and Lysobacter (Pseudomonadota) were most abundant in lower WV permafrost only. Within Firmicutes, the most abundant genus was Clostridium sensu stricto 9, an anaerobic, spore-forming bacteria. The genus Conexibacter was abundant in both the surface and subsurface at lower WV.
The most abundant phyla for the PV subsurface permafrost sample (PV2) were Actinobacteriota (40.9%), Proteobacteria (18.9%), Chloroflexi (17.0%), and Verrucomicrobiota (8.5%). For the PV subsurface permafrost, the most abundant ASVs were within families Chthoniobacteraceae, Cyclobacteriaceae, Nitrospiraceae, and Nocardioidaceae. Nitrospira (Nitrospirota) were most abundant in PV permafrost only. The most abundant ASVs were within the genera Nocardioides, Candidatus Udaeobacter, Gaiella, Conexibacter, Sphingomonas, and Nitrospira. The genera Conexibacter, Candidatus Udaeobacter, and Sphingomonas were abundant in both the surface soil and subsurface permafrost at PV.
Potential metabolic characteristics in subsurface permafrost at both lower WV and PV include chemoorganotrophy (Nocardioides) and oligotrophy (Blastocatella, Candidatus Udaeobacter). Potential metabolic characteristics detected in lower WV permafrost include fermentation (Clostridium sensu stricto 9) and high-temperature sulfate reduction (Desulfobacterota). Potential metabolic characteristics detected in PV permafrost include chemoorganotrophy (Gaiella), chemolithoautotrophy, and nitrite oxidation (Nitrospira). The abundance of Nocardioides, Blastocatella, Sphingomonas, and Candidatus Udaeobacter demonstrates the subsurface ice-cemented permafrost is dominated by aerobic heterotrophs and oligotrophic conditions. At both lower WV and PV, methanogens were detected in the subsurface permafrost. No methanogens were detected in the surface soils at either site. Two genera of methanogens within the phylum Halobacterota (Methanosphaerula and Methanosarcina) were detected in the lower WV permafrost core, and they comprise <0.03% of relative abundance. In the PV permafrost core, Methanosarcina was detected and comprises 0.02% of the relative abundance.
The lower WV and PV permafrost cores both showed greater alpha diversity in comparison with the corresponding surface soils (Fig. 4). The comparison revealed Shannon diversity was highest in the lower WV permafrost core. Both sites showed that the subsurface permafrost was more diverse than the surface soil.
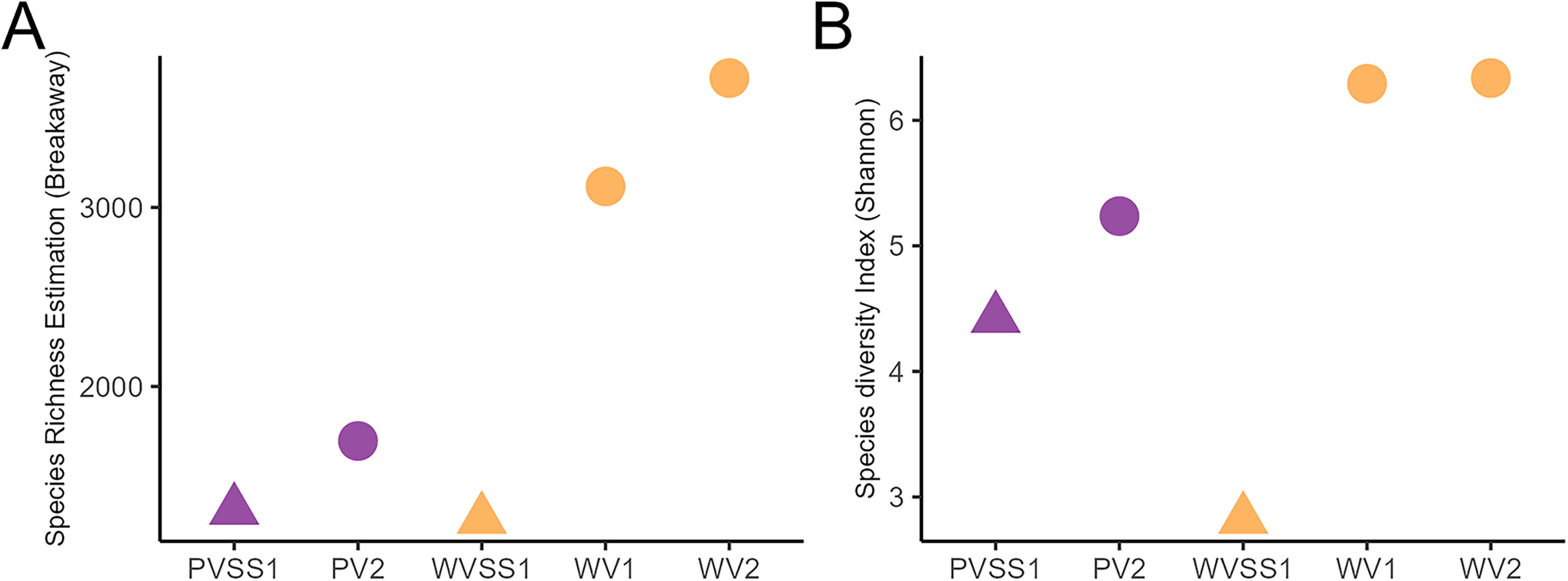
Alpha diversity plot showing Breakaway
Principal coordinate analysis revealed that the structure of the SSs at lower WV and PV was more similar to each other than each sample was to their respective subsurface permafrost sample (Fig. 5). Figure 6 shows the community compositional dissimilarity (beta diversity) of surface soil and subsurface permafrost samples at lower WV and PV, and the dry surface soil samples located at Lake Bonney, Lake Brownworth, and Lake Vanda (from Monteiro et al., 2021). The SSs at lower WV and PV were broadly consistent with the dry samples near Lake Brownworth and Lake Vanda (Monteiro et al., 2021). The subsurface permafrost samples from both sites varied significantly from the SSs. At lower WV and PV (WV1, WV2, and PV2), microbial communities in permafrost had more shared connections than the subsurface permafrost and surface soil at each site (e.g., WV1 and WVSS1).
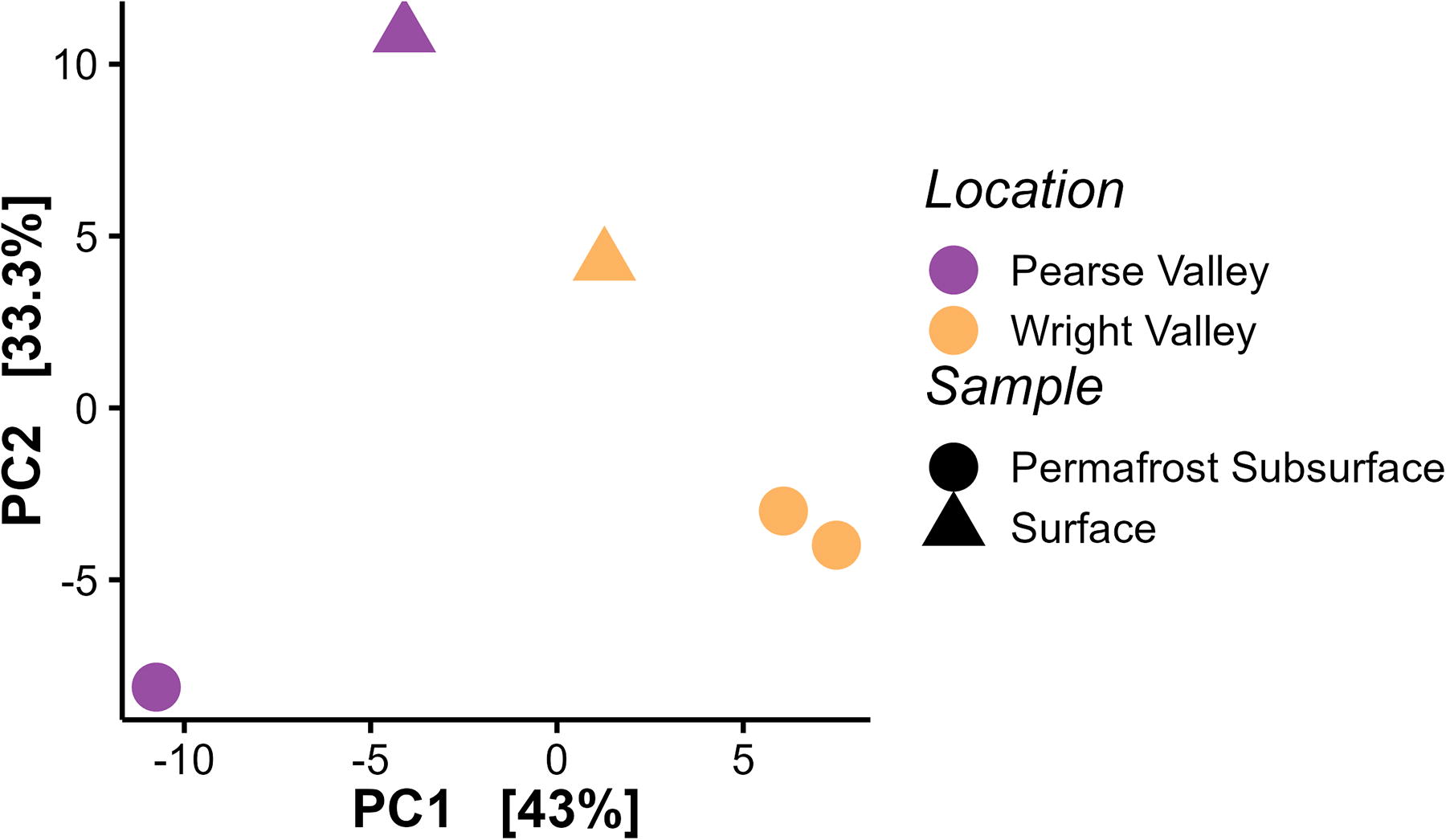
Principal coordinate analysis beta diversity plot of surface soil and subsurface permafrost samples from lower Wright Valley and Pearse Valley.
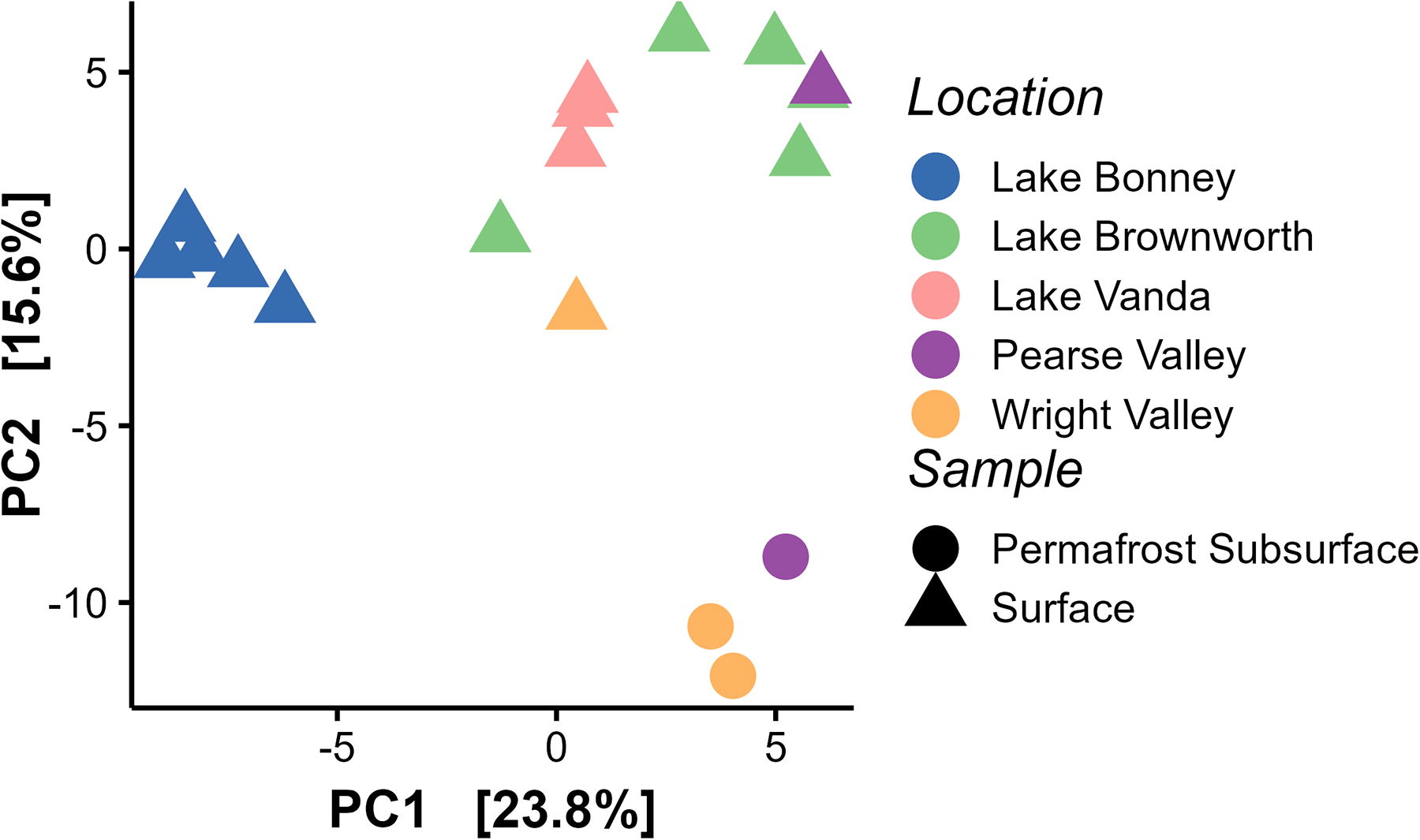
Principal coordinate analysis of microbial composition from surface and subsurface samples at lower Wright Valley and Pearse Valley, and dry surface samples located at Lake Bonney, Lake Brownworth, and Lake Vanda (from Monteiro et al., 2021). The dry Lake Bonney samples had a relatively high abundance of cyanobacteria, pulling the samples across PC1.
Microbial communities in surface soil and subsurface permafrost
Both SS and subsurface sample were dominated by the phyla Acidobacteriota, Actinobacteriota, and Bacteroidota, which are commonly found in McMurdo Dry Valley soils (e.g., Cary et al., 2010; Chan-Yam et al., 2019; Monteiro et al., 2021). Comparison of the relative abundance of bacteria at both lower WV and PV sites showed that Chloroflexi, Firmicutes, Proteobacteria, and Verrucomicrobiota were at greater abundance in subsurface permafrost than in surface soil samples. Chloroflexi and Firmicutes were also found to be more abundant in deeper soils in Alaskan permafrost (Deng et al., 2015), whereas Bacteroidota were more abundant in surface soils than in subsurface permafrost at both sites. The greater proportion of Bacteroidota in upper surface soils and Proteobacteria in deeper soils has previously been observed in PV (Chan-Yam et al., 2019). Proteobacteria, Firmicutes, Bacteroidota, and Actinobacteria also dominated the bacterial community in University Valley permafrost (Goordial et al., 2016). However, the abundance of these phyla in all samples is not a useful indicator; rather, it is those individual taxa (at the genus or ASV level) whose abundance varies between samples that are required to determine differences in the depositional environment and paleohabitat.
ASVs within both lower WV and PV permafrost cores were dominated by organisms that are typically aerobic heterotrophs. Nocardioides, Gaiella, and Luteitalea are abundant in subsurface permafrost at both sites, but not in the surface soil. Nocardioides are chemoorganotrophic, aerobic bacteria (Tóth and Borsodi, 2014) and are consumers of organic matter (Gesheva and Vasileva-Tonkova, 2012). Rare and little known parasitic Dependentiae was identified in both permafrost cores. In PV, microbial communities of soils identified using 16S rRNA gene sequencing (Chan-Yam et al., 2019) were dominated by Actinobacteria, Bacteroidetes, Chloroflexi, and Proteobacteria, as was seen in our study.
Culturing of samples from University Valley (Goordial et al., 2016) also cultivated isolates from Sphingomonas, which is abundant in the lower WV and PV cores, and the nitrite oxidizer Nitrospira, which is abundant in PV. Culturing of samples from Beacon Valley (Gilichinsky et al., 2007) also cultivated isolates from Sphingomonas and Nocardioides, which were abundant in the lower WV and PV cores.
At lower WV, the permafrost has been isolated from the overlying active layer surface soil for >7000 years and at PV for >180,000 years (Anderson et al., 2023). Thus, subsurface permafrost at these two sites has been permanently frozen and isolated from the cryoturbated surface soil for an extended period and has an abundance of bacteria that likely favor oligotrophic conditions. While microorganisms from lower WV and PV are not in permafrost as old as cultured microbial populations from high-elevation Antarctic permafrost environments (>5 Ma) (Gilichinsky et al., 2007), they demonstrate that 16S rRNA gene amplicon sequencing can be used to identify microbes in Pleistocene subsurface ice-cemented permafrost but may not be suitable in high-elevation Miocene ice-cemented permafrost, such as the Friis Hills. The presence of anaerobic microorganisms (e.g., sulfate reducers and methanogens) that are unlikely to be introduced from the air, at both sites, also provides further evidence of extant or ancient microbial communities that are isolated from the surface.
The occurrence of methanogens in subsurface permafrost at lower WV and PV may indicate that the sediments were of lake origin. Methanogens detected in 15,000-year-old permafrost sediments from Miers Valley are interpreted to be associated with Glacial Lake Trowbridge, a large lake that occupied Miers Valley between 10,000 and 23,000 years ago (Vishnivetskaya et al., 2018).
Microbial communities from cores at both lower WV and PV sites were more similar to those in permafrost from each respective valley than they were to their overlying surface soils. The relatively high diversity of microbial communities in subsurface permafrost in lower WV and PV could reflect well-preserved ancient DNA or extant taxa persisting in thin films of brine, enveloping sediments, which could help microbes remain dormant in these permafrost habitats (Gilichinsky et al., 1993; Gilichinsky and Wagener, 1995). Spore-forming bacteria (e.g., clostridia) may also be more abundant with depth and age due to genes involved in dormancy (Mackelprang et al., 2017). Our analyses suggest that subsurface permafrost habitats are either more suitable for many microbial taxa than harsh, dry surface soils or that DNA is better preserved in the subsurface compared with the surface, which experiences higher exposure to UV and desiccation and thus greater degradation.
We report higher diversity in subsurface permafrost than SS. While low-biomass samples are reported to have inflated diversity metrics due to contaminants (Karstens et al., 2019), the results from our decontamination protocol demonstrate that diversity remains higher in subsurface than in SS even with the removal of contaminants. Microbial diversity may also be inflated in low-biomass samples due to higher proportions of fragmented DNA that interfere with lower proportions of extant cells, which may lead to inflated diversity metrics. If the DNA is not ancient, an alternative hypothesis is that higher diversity in permafrost may be associated with an extant community surviving within brine channels that has diversified over time, associated with the duration of stability and isolation, free of external influence (Mackelprang et al., 2017). Surface soils, in contrast, are dynamic and influenced by external factors such as katabatic winds, aeolian sedimentation, cryoturbation, and snowfall. Processes that influence soil disturbance and stability may influence the diversity and composition of the microbial community. Higher diversity of microbial communities has also been shown in low input, stable conditions such as the deep sea (Jacob et al., 2013; Li et al., 2009; Schauer et al., 2010) and at New Harbour, in the Ross Sea (Currie et al., 2021). Such stable conditions may select for more diverse and specialized microbial taxa (Dayton and Oliver, 1977; Currie et al., 2021). However, these published low-biomass communities may also have inflated diversity metrics.
If microbial populations can be identified and be reliably constrained by age in Antarctic permafrost, they may reflect past climatic and environmental conditions, different from those of the present surface, prior to the landscape being frozen, or they may reflect changes in metabolism over geologic time. It has been suggested that microbial communities in Alaskan permafrost do not reflect the climate and vegetation at the time the permafrost formed (Mackelprang et al., 2017). Instead, once frozen, microbes respond to subzero temperatures, rather than surface conditions. Here, we suggest the isolation of a habitat and stability over time record paleoecology of the surface habitat during the time of freezing, which is different from the present-day surface. Our study demonstrates the potential for paleoclimate records preserved within permafrost, to be reconstructed from microbial paleoecology. Using this approach, it may be possible to recover a complete Pleistocene microbial assemblage, which could be compared and correlated with strata from other permafrost horizons.
The low abundance and diversity of Archaea in surface soil and subsurface permafrost in our work has also been reported in previous studies from the McMurdo Dry Valleys (Khan et al., 2011; Bates et al., 2011; Goordial et al., 2016, 2017). Other Dry Valleys studies found Archaea absent from all niches (Pointing et al., 2009). To date, archaeal diversity and associated ecosystem processes in the McMurdo Dry Valleys remain poorly understood (Khan et al., 2011; Bates et al., 2011). In our study, the predominance of Crenarchaeota is consistent with findings in Miers Valley (Khan et al., 2011) and Victoria Valley (Ayton et al., 2010) surface soils. The presence of Methanomicrobia in the lower WV permafrost core has also been detected in surface soil samples at University Valley (Goordial et al., 2016).
Decontamination protocol
Our pGEM decontamination protocol is a novel approach to verify the presence of isolated microbial DNA in low-biomass permafrost core samples. The method used to decontaminate the exterior of the permafrost cores is similar to techniques that have previously been used successfully on protocols for ice cores (e.g., Christner et al., 2005; Rogers et al., 2004; Zhong et al., 2021) and permafrost (e.g., Bang-Andreasen et al., 2017), but with the addition of using wash baths. In addition, we applied sonication to detach loosely bound negatively charged DNA from positively charged sedimentary particles (Supplementary Fig. 3). This was followed by sampling of both water and fine sediments using a peristaltic pump (Supplementary Fig. 4). This protocol may assist future researchers who investigate ultra-low-biomass samples and the limits of life, in particular when there is concern with contaminants, while additionally competing with sediments that bind to DNA and reduce extraction efficiency.
While our field sampling, decontamination protocol, DNA extraction, and sample processing steps followed stringent procedures to prevent and detect contamination prior to sequencing, our bioinformatic analysis resolved the presence of potential contaminants in the control samples. These contaminants may have been introduced during the sequencing step or may have been present throughout the entire quality control process due to detection limits of DNA quantification and qPCR. For example, the DNA concentration detection limit for Qubit HS is 0.005 ng/μL. A single bacterial cell is estimated to contain 0.5–20 fg of DNA, which is orders of magnitude lower than the detection limit of the Qubit Fluorometer dsDNA HS assay kit. The difference in cell numbers and detection approach can lead to false-negative results and is an important consideration when working with low-biomass samples where low cell counts can be a limiting factor. In comparison, qPCR is a robust quality control technique that can detect gene presence to one gene copy with 95% probability (Bustin et al., 2009). A limit to the sensitivity of qPCR single-copy detection can occur when the amplified gene target is mixed within a pool of primer dimers. This generates a qPCR signal that is undifferentiable from negative controls and is resolved with the addition of a high-resolution melt step after qPCR amplification. We applied a high-resolution melt in this study and did not identify any signatures of pGEM contamination in the samples or the controls. A second limitation of qPCR that may be specific to our study is that amplification of a target gene depends on the specific primer binding sites. If these targeted locations are fragmented, which is prevalent in ancient DNA, then variability may be introduced into the first three cycles of PCR and may impact the detection of single gene copies. Despite the stringent steps taken in our study to detect contaminants prior to sequencing, we suggest that the sequencing depth (>400,000 reads) may explain why we identified low-level contaminants, namely, Pseudomonas, in the extraction blanks. These sequences may remain undetected when shallower sequencing approaches are applied (e.g., 10,000 reads). For low-biomass samples, sequencing to a depth of 100,000–200,000 reads may enable the differentiation between ultra-low-biomass samples and those environments where DNA is no longer present in long enough consecutive sequence to be amplified via PCR. In these situations, metagenome sequencing of low-biomass ancient DNA would be preferential. We acknowledge that the development of a standardized sequencing depth for low biomass would require further testing. It is important to note that our samples are representative of taxa that are novel to the contaminants identified in the controls. This distinction supports our approach that the sequenced microbial communities reflect the true microbial diversity of surface soil and subsurface permafrost.
DNA was detected from samples from the Friis Hills; however, the low number of reads (three to nine) post deep sequencing suggests that the DNA is likely to be highly fragmented and not amplifiable via techniques such as PCR that rely on targeting specific intact sequences (Johnson et al., 2007). This observation is consistent with the stability of permafrost since the late Miocene (Verret et al., 2023) and perhaps the preservation of such ancient DNA. The inability to identify DNA using amplicon sequencing at the Friis Hills further validates the effectiveness of the decontamination protocol in permafrost. Since no amplicons could be resolved, it suggests that any potential contaminants were effectively removed during decontamination procedures. This strengthens the reliability of the results from the permafrost cores at lower WV and PV where DNA was identified and ensures reliable microbial community analyses.
Habitability in the stable upland zone
Using 16S rRNA gene amplicon, our method was unable to characterize the microbial community in surface soil or subsurface ice-cemented permafrost from the Friis Hills, perhaps due to high levels of DNA fragmentation or the absence of DNA at this site. These results are consistent with some previous studies (Dragone et al., 2021, 2022) but inconsistent with others (Goordial et al., 2016, 2017), which suggests that microbial communities in the coldest, driest, high-elevation (>1000 masl) soils are severely restricted. While our studies have not been able to identify taxa from high-elevation stable upland zone sites using 16S rRNA approaches, in the inland mixed zone, our approach found diverse microbial communities are present in >180,000-year-old ice-cemented permafrost. Thus, our results suggest there may be a limitation to detecting life via DNA assays in hyperarid, high-elevation Antarctic environments, or a microbiological “treeline” exists >1000 m elevation in the Transantarctic Mountains.
The challenges of detecting microbial life at high elevations suggest that temperature gradients and aridity significantly influence microbial habitability or DNA preservation. The negligible difference in mean annual air temperature between the coastal thaw and stable upland zone of ∼2°C (Obryk et al., 2020) is due to the warming effect of katabatic winds (Doran et al., 2002). However, the mean annual ground temperature at 10 cm depth may be more relevant for understanding the relationship between microbial communities and permafrost. For example, the difference in mean annual ground temperature between lower WV at −19.4°C (Obryk et al., 2020) and University Valley at −24.2°C (Goordial et al., 2016) is ∼5°C. When the elevation change from 280 m at lower WV to 1677 m at University Valley, which is 381 m higher than the FHDP-2B drill site, is taken into account, this temperature difference may also be considered negligible. These observations demonstrate that there is no clear relationship between mean annual temperature and elevation or distance from the coast in the McMurdo Dry Valleys. However, during summer months (December, January, and February), when there is a reduction of katabatic winds, the average air temperatures vary as expected with the adiabatic lapse rate (9.8°C km−1) and distance from the coast (Doran et al., 2002). Based on these observations, we speculate that the lack of days above freezing during summer in the stable upland zone, compared with the inland mixed and coastal thaw zones, limits water availability for heterotrophic communities at high elevations, and the maximum temperatures of the ice table during summer may be more important than the mean annual temperatures.
Implications for habitability on Mars
Understanding temperature gradients and water availability in the McMurdo Dry Valleys provides valuable context for future site selections when investigating the habitability of martian permafrost. During periods of high obliquity on Mars in the recent (10 Ma) geological past, insolation on polar-facing slopes in the mid and high latitudes was higher (de Haas et al., 2015; Laskar et al., 2002; Marchant and Head, 2007; Head et al., 2008). The high obliquity allows for seasonal, insolation-driven surface melting of snow and ice, and wetter climate conditions. For example, surface melting could have occurred on the northern polar plains of Mars ∼5 million years ago (McKay et al., 2013). Therefore, permafrost conditions at stable upland zone sites such as University Valley (McKay et al., 2017) and inland mixed zone sites such as PV (Marchant and Head, 2007) may provide useful analogs for temperature gradients during these warmer and wetter periods on Mars. During warmer climate conditions at the north and south poles of Mars, insolation exceeds the threshold for active layer formation, and seasonal active layers may have formed (Marchant and Head, 2007). The preservation of buried snowpack deposits in PV may also function as an analog for deposits in the north polar erg of Mars and act as water sources in hyperarid environments (Heldmann et al., 2012).
The periods of high obliquity on Mars could have produced subsurface temperatures above −20°C, allowing the formation of thin brine films, and microbial activity may have been possible (McKay et al., 2013; Rivkina et al., 2000). Therefore, subsurface chemoautotrophy could be a viable survival strategy in martian permafrost (McKay et al., 2013). Nitrogen is present in the atmosphere at low levels, and nitrates may be present in the martian soil and could be directly usable by microorganisms (McKay et al., 2013). We observed similar metabolic characteristics in the PV permafrost core, where nitrite-oxidizing bacteria, such as Nitrospira, are abundant.
The age and long-term stability of high-elevation permafrost in Antarctica (e.g., Anderson et al., 2025; Bergelin et al., 2022; Verret et al., 2023) may be comparable to the estimated age of the seasonal active layer wetting and surface evolution of permafrost in the northern polar plains of Mars. This provides the best analog for near-surface martian permafrost. Considering the challenges of contamination and sequencing DNA of ultra-low-biomass samples, the development of sampling strategies and analyses to detect life in the stable upland zone of the McMurdo Dry Valleys has implications for the extremities of life and is directly relevant to searching for habitability on Mars.
Conclusions
We have developed a new decontamination protocol and low-biomass extraction approach to identify microbial populations from ice-cemented permafrost in lower WV and PV in the McMurdo Dry Valleys of Antarctica. At both sites, the analyses revealed that the subsurface permafrost exhibited higher microbial diversity compared with the surface soil. This technique was unable to characterize microbial communities from a high-elevation stable upland zone site, Friis Hills, which is consistent with some previous efforts (Dragone et al., 2021, 2022) though inconsistent with other efforts (Goordial et al., 2016, 2017) to analyze microbial communities in high-elevation soils and permafrost using 16S rRNA gene amplicon sequencing. This suggests microbial preservation or dormancy is severely restricted in persistent cold, arid habitats. The analysis of microbial communities in ice-cemented permafrost compared with surface soil from lower WV and PV revealed diverse characteristics consistent with an ancient and isolated microbial community. Microorganisms in permafrost are isolated from the surface soil, and perhaps cold-temperature adaptations, slow metabolism, and evolution enable communities to maintain viability over multiple glacial–interglacial cycles. The absence of bacterial taxa such as Clostridiaceae and genera such as Nitrospira and Gaiella in the surface soil suggest that these subsurface microbial communities have been isolated from the surface for a long time or that these are two distinct ecological niches for microorganisms. These findings provide insight into the diversity of microbial life in permafrost and may represent a powerful indicator for paleoecological reconstruction where other fossils are rare or nonexistent. Our results further demonstrate that high-elevation Antarctic permafrost in the stable upland zone nears the environmental limits of life. Thus, the study of microbial life in ice-cemented permafrost in the McMurdo Dry Valleys represents the closest terrestrial analog to martian permafrost. Based on this understanding, to search for possible life on Mars, it may first be necessary to develop tools to detect life reliably in the stable upland zone of the McMurdo Dry Valleys.
Footnotes
Acknowledgments
The authors thank Bob Dagg and Steph Lambie for assistance in the field, Antarctica New Zealand and Southern Lakes Helicopters for logistical support. They thank Tim Naish, Richard Levy, and Webster Drilling and Exploration (Tony Kingan and Adam Rutland) for assistance in collecting the Friis Hills samples. Chris McKay and an anonymous reviewer are thanked for their helpful recommendations that greatly improved the article.
Authors’ Contributions
J.T.H.A., G.S.W., S.C.C., I.R.M., A.A., and N.D. conducted the field work and sample collection. R.R.-B., J.T.H.A., A.J.M., and S.C.C. conducted the laboratory work. A.J.M., J.T.H.A., and S.C.C. performed the data analysis. J.T.H.A. prepared the article with contributions from all authors.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the New Zealand Antarctic Research Institute (RFP 2014-1). J.T.H.A. was supported by a Sir Robin Irvine Scholarship and a University of Otago departmental award. A.J.M. was supported by the Rutherford Foundation Royal Society Te Aparangi Postdoctoral Fellowship (20-UOW006) and the Antarctic Science Platform Opportunities Fund (ASP-027-02). A.A. and N.D. were partially supported by the Russian Antarctic Expedition.
Supplementary Material
Supplementary Data S1
Supplementary Data S2
Associate Editor: Radu Popa
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.