
Abstract
Keywords
Introduction
In 2010, an estimated 202 million people were living with peripheral artery disease (PAD) worldwide. 1 However, limited data are available on the prevalence of PAD in China, with published reports suggesting rates ranging from 2.5% to 20%, varying between urban and rural areas.2–5
Over the decades, the original endovascular treatment for symptomatic lower limb PAD, percutaneous transluminal angioplasty (PTA), was supplanted by bare metal and drug-eluting stents (DES). However, their efficacy has been limited in complex lesions, whether they are long, heavily calcified, or occurring in areas already treated by a stent. In addition, the long-term consequences of a permanent metallic implant and stent fracture remain unknown.6,7
Drug-coated balloons (DCBs) were developed to overcome the challenges of PTA and stenting, particularly in the superficial femoral (SFA) and popliteal arteries. Randomized controlled trials (RCTs) have shown DCBs to be both safe and effective for the treatment of lesions in the femoropopliteal segment.8–14 These trials, however, have been composed almost entirely of Caucasian subjects. While several DCB trials are ongoing in Asian populations [LTX DCB China (NCT02720003), Ranger China (NCT029440710), Biolux P4 China (NCT02912715), and Ranger Japan (NCT03064126)], there is still a paucity of published clinical results in Asian populations. Only the AcoArt I study15,16 and the MDT-2113 (IN.PACT) SFA Japan trial17,18 have been published through late 2018. The 12-month results of the IN.PACT SFA China study are now presented.
Materials and Methods
Study Design
The IN.PACT SFA Clinical Study for the Treatment of Atherosclerotic Lesions in the SFA and/or Proximal Popliteal Artery using the IN.PACT Admiral Drug-Eluting Balloon in a Chinese Patient Population was an independently-adjudicated, prospective, multicenter, single-arm, premarket study designed to confirm the safety and effectiveness of the IN.PACT Admiral DCB in Chinese subjects. The 15 participating sites and principal investigators are listed in Supplemental Table 1 (available in the online version of the article). After completion of the clinical trial, the product name was updated to IN.PACT Admiral Paclitaxel-Coated PTA Balloon Catheter, though it will be referred to as the IN.PACT Admiral DCB in this report. The IN.PACT Admiral DCB is coated with the antiproliferative agent paclitaxel (3.5 µg/mm2) in a urea excipient.
As an open label study, blinding procedures were not applicable. An independent clinical events committee adjudicated major adverse events. Duplex ultrasonography studies were reviewed by an independent vascular ultrasound core laboratory (VasCore, Massachusetts General Hospital, Boston, MA, USA), while angiograms were evaluated by the Beth Israel Deaconess Imaging Laboratory (Beth Israel Deaconess Medical Center, Boston, MA, USA). This study was conducted in compliance with the 2008 version of the Declaration of Helsinki, the international standard ISO 14155:2011 (“Clinical Investigation of medical devices for human subjects”), and applicable China laws and regulations. Prior to enrolling subjects in this study, the ethics committee or institutional review board at each site approved the study protocol. This study is registered on the National Institutes of Health website (ClinicalTrials.gov; identifier NCT02118532).
Inclusion and Exclusion Criteria
Patients were eligible for enrollment if they were between the ages of 18 and 85 years, had a documented diagnosis of symptomatic lower limb PAD classified as Rutherford category 2 to 4 claudication and/or rest pain, and had a life expectancy >12 months. Target lesions for inclusion were de novo or native artery restenotic lesions in the SFA and/or proximal popliteal artery. Total occlusions had to be ≤10 cm in length, while lesions with a diameter stenosis ≥70% and <100% could have a total lesion length between 4 and 20 cm. Patients had to have at least 1 runoff vessel to the foot, and any iliac lesions had to be treated with approved devices before the target lesion. After signing an informed consent form, subjects were enrolled in the study following lesion crossing and successful predilation.
Subjects were excluded from the trial if they had a stroke or myocardial infarction within 3 months prior to the index procedure; known allergies or hypersensitivities to components of the procedure (heparin, aspirin, contrast media, paclitaxel); or surgeries that took place within 30 days prior to or scheduled after the procedure. Angiographic exclusion criteria included aneurysm, thrombus, severe calcium, in-stent restenosis, failure to cross the target lesion, a grade D or higher flow-limiting dissection, or residual stenosis >70% after predilation. A full list of inclusion and exclusion criteria is available on the ClinicalTrials.gov website.
Treatment, Medical Therapy, and Follow-up
In this trial, there was no mandatory medication prescribed, although dual antiplatelet therapy was recommended. In general, a minimum of 75 mg of clopidogrel was given daily for at least 3 days prior to the index procedure; if this was not feasible, a 300-mg bolus of clopidogrel was administered within 24 hours prior to the procedure (or a 500-mg ticlopidine bolus if allergic to clopidogrel). Prior to the procedure, a 500-mg loading dose of aspirin was given if not already on a 75 mg/d regimen. As appropriate, heparin was administered during the procedure to maintain an activated clotting time ≥250 seconds. Additional information is provided in Supplemental Table 2 (available in the online version of the article).
Lesions were predilated with a standard uncoated PTA balloon sized 1 mm smaller than the reference vessel diameter (RVD). After successful predilation, an IN.PACT Admiral DCB sized to match the RVD and slightly longer than the lesion length was delivered to the target site. The DCB was inflated across the lesion at or beyond nominal pressure for at least 3 minutes. More than one DCB could be used in the study to treat a given lesion. If there was a residual stenosis >50%, a translesion gradient >10 mmHg, and/or a flow-limiting dissection (grade D or higher) then postdilation was performed with a PTA balloon shorter than the full lesion length. If postdilation was unsuccessful even with a long inflation time of ≥3 minutes, provisional stenting was allowed, but not with a DES.
Subjects were followed by their treating physician at 30 days, 6 months, and 12 months. These office visits included duplex ultrasound and assessment of adverse events. Functional testing included the Walking Impairment Questionnaire (WIQ), 6-minute walk test (6MWT), and EuroQol-5 dimensional quality of life test (EQ-5D). If a reintervention was required, PTA and provisional stenting were used.
Qualitative and Quantitative Angiography
Procedure angiograms were independently reviewed by observers blinded to the clinical outcomes. Lesion length was defined as the shoulder-to-shoulder lumen narrowing that was to be treated. RVD and percent diameter stenosis were determined using quantitative angiographic methods (CMS Medis, Leiden, the Netherlands). Dissections were graded using the National Heart, Lung and Blood Institute grading system. 19
Enrolled Subjects
Between March 2014 and August 2015, 143 subjects (mean age 66.8±7.7 years; 107 men) were enrolled at 15 centers in China. Patient flow through 12-month follow-up is shown in Figure 1. Patient characteristics are listed in Table 1. Diabetes mellitus was highly prevalent (66, 46.2%), a third of the subjects were smokers (52, 36.4%). The majority of subjects were categorized as Rutherford category 2 and 3 [69 (48.3%) and 55 (38.5%), respectively]; 19 (13.3%) subjects had critical limb ischemia (CLI; Rutherford category 4). Mean lesion length was 10.4±6.51 cm, and 17 (11.9%) lesions were severely calcified as determined by the angiographic core laboratory. All lesions except one were de novo. More than half the lesions were occlusions (75, 52.4%), and the mean diameter stenosis was 89.0%.
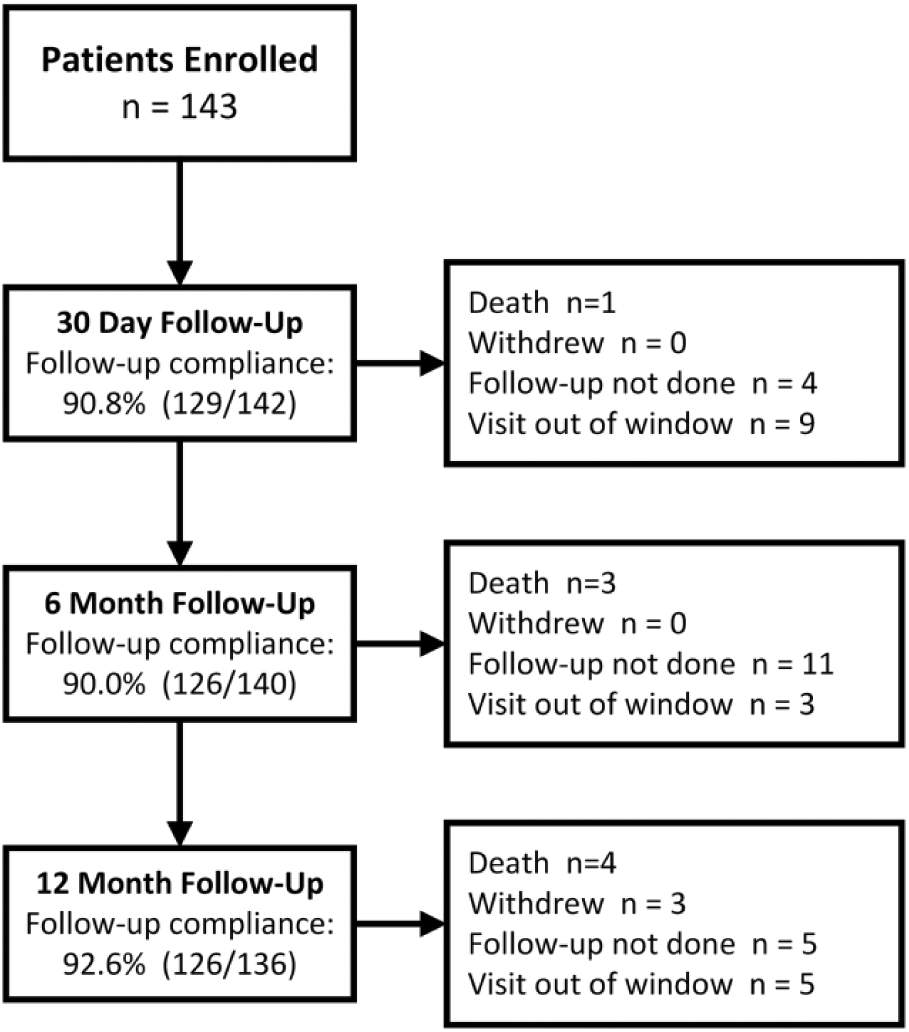
Enrollment in the IN.PACT SFA China study, showing deaths, subjects lost to follow-up, visits not completed, and withdrawals through 12 months.
Baseline Demographic and Lesion Characteristics of the 143 Study Subjects. a
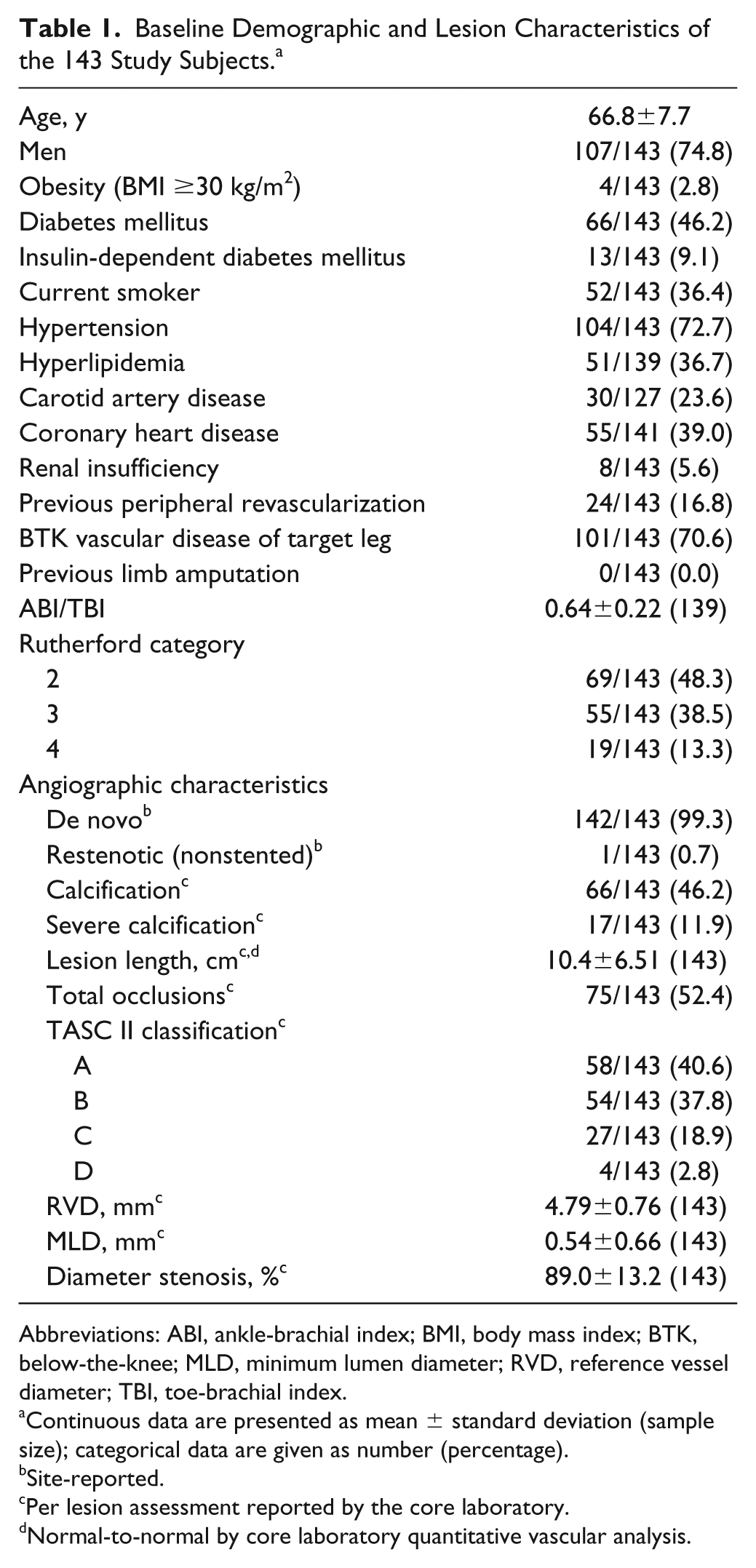
Abbreviations: ABI, ankle-brachial index; BMI, body mass index; BTK, below-the-knee; MLD, minimum lumen diameter; RVD, reference vessel diameter; TBI, toe-brachial index.
Continuous data are presented as mean ± standard deviation (sample size); categorical data are given as number (percentage).
Site-reported.
Per lesion assessment reported by the core laboratory.
Normal-to-normal by core laboratory quantitative vascular analysis.
Study Outcomes
The primary effectiveness outcome was primary patency within 12 months of the index procedure, defined as freedom from clinically-driven target lesion revascularization (CD-TLR) and freedom from restenosis determined by a duplex ultrasound peak systolic velocity ratio ≤2.4. This endpoint was evaluated against a performance goal of 50% 20 in all subjects who did not receive a stent [intent-to-treat (ITT) population]. This number was generated when the IN.PACT China study was designed in 2013. Eleven clinical trials21–31 examining DCB, DES, bare metal stents, and PTA were assessed, and the treatment differences between the non-DES devices with and without provisional stenting were evaluated. To assess the 12-month primary patency performance in DCB and PTA separately, the published data were combined, both unweighted and as a weighted average of the individually calculated 12-month rates of primary patency, TLR, and binary restenosis. The unweighted rate calculated the proportion of the number of patients with the event over number of all participating patients across the studies (raw count). The weighted average’s weights depended on variation in sample size (meta-analytic rate).32,33 The final expected performance in DCB and PTA also accounted for the difference between devices used with a stent and without a stent, as the primary analysis was planned in subjects who did not receive a provisional stent.
The primary safety outcome was a composite of freedom from device- and procedure-related mortality, major target limb amputation, and CD-TLR within 30 days after the index procedure. The performance goal for this outcome was 88%. 20 Secondary outcomes included major adverse events (MAE) through 12 months, death, CD-TLR, clinically driven target vessel revascularization (CD-TVR), device success, and procedure success. Device success was defined as successful delivery, inflation, deflation, and retrieval of the intact study balloon without burst (less than burst pressure). Procedure success was residual stenosis ≤50% for non-stented subjects or ≤30% for stented subjects. Clinical success was procedure success without complications (death, major target limb amputation, thrombosis of the target lesion, or TVR) prior to discharge. Primary sustained clinical improvement was defined as a sustained upward shift of at least 1 Rutherford category compared with baseline without the need for endovascular or surgical TLR in amputation-free survivors.
Statistical Methods
All analyses were based on the intention-to-treat principle except for the primary effectiveness outcome; subjects with provisional stenting were not included. All summaries were based on subjects or lesions with evaluable data. No imputation was performed for missing data. For baseline characteristics, continuous variables were described as mean ± standard deviation; dichotomous and categorical variables were described as counts and proportions. The Kaplan-Meier method was used to evaluate time-to-event data for primary patency and freedom from CD-TLR over the 12-month follow-up period, including all ITT subjects. Estimates are given with the 95% confidence intervals (CI). For event rates that were expressed as proportions, the number of subjects with an event was the numerator and the total number of subjects with either an event or at least 330 days of clinical follow-up was the denominator. For assessment of clinical characteristics at 12 months, subjects were required to have data at both baseline and 12 months but were not required to have a full 330 days of clinical follow-up. Statistical analyses were performed using SAS software (version 9.4; SAS Institute, Cary, NC, USA).
Results
While all lesions were predilated, postdilation was performed in only 20 (14.0%) subjects (Table 2). Device success was achieved in 97.6% of subjects (206/211 balloons used). Flow-limiting dissections (all grade D) occurred in 37 (25.9%) subjects; despite this, the rate of provisional stenting was 4.2% (6/143).
Characteristics of the Procedures in 143 Study Subjects. a
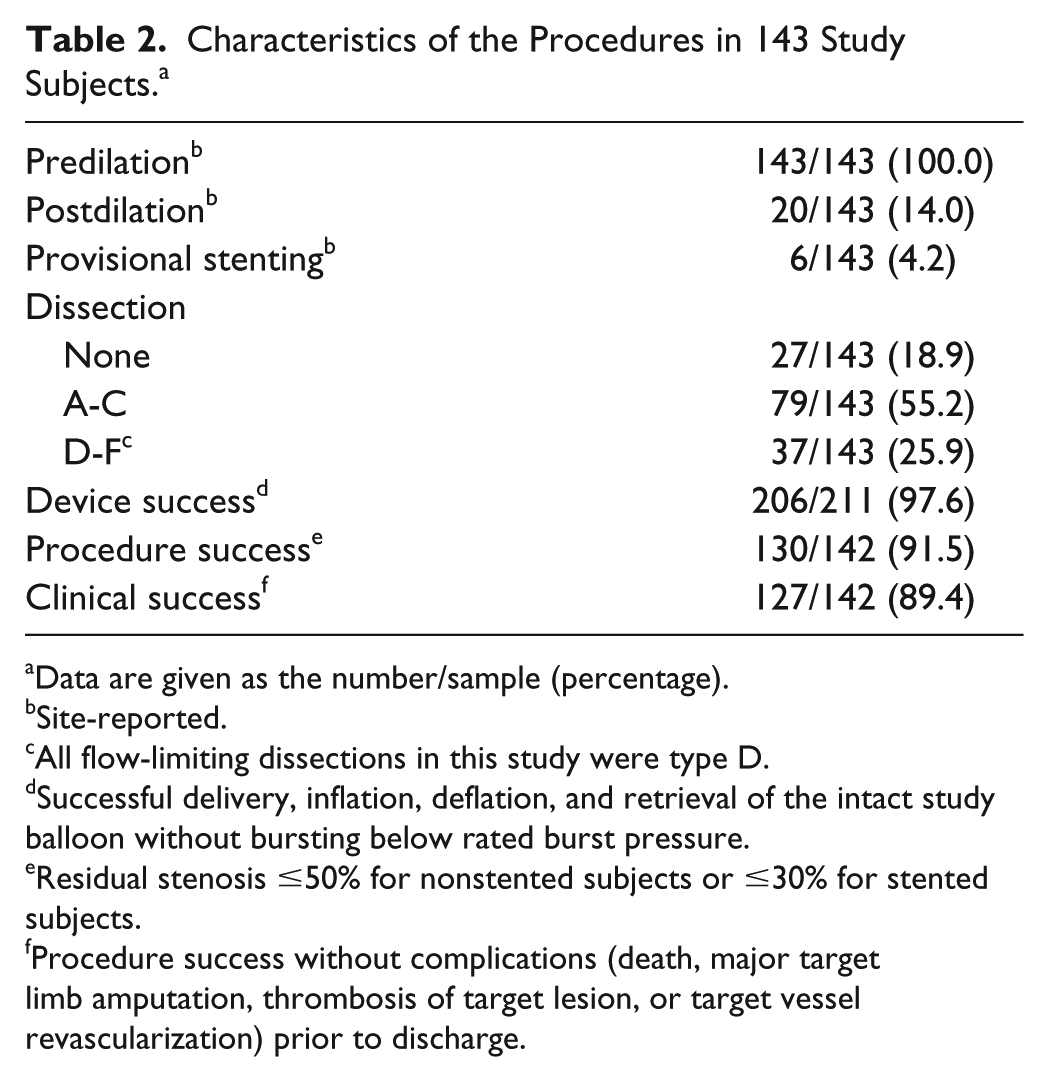
Data are given as the number/sample (percentage).
Site-reported.
All flow-limiting dissections in this study were type D.
Successful delivery, inflation, deflation, and retrieval of the intact study balloon without bursting below rated burst pressure.
Residual stenosis ≤50% for nonstented subjects or ≤30% for stented subjects.
Procedure success without complications (death, major target limb amputation, thrombosis of target lesion, or target vessel revascularization) prior to discharge.
Efficacy Outcomes
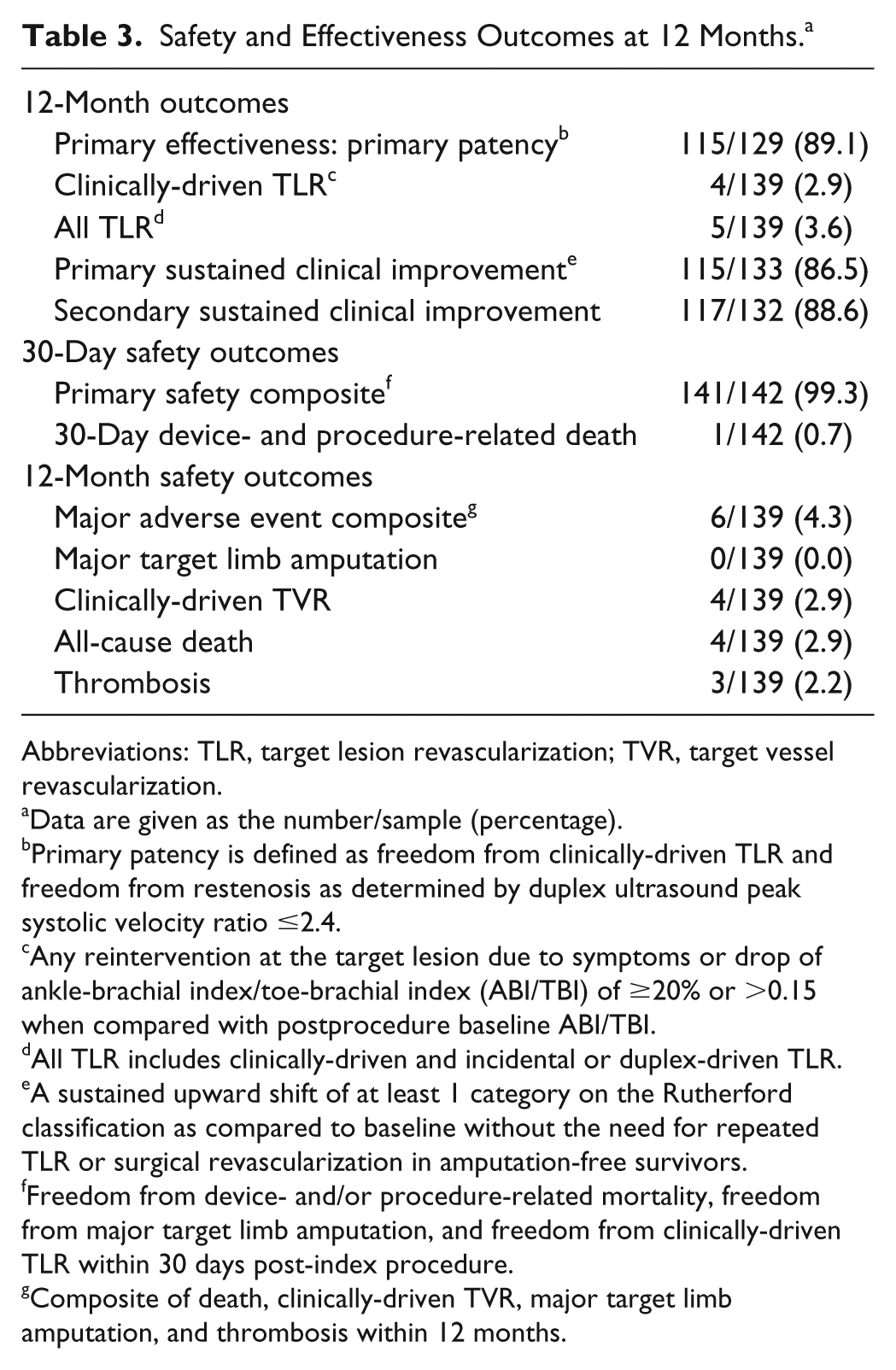
Follow-up compliance at 12 months was 92.6% (126/136). The 1-year primary patency of DCBs for nonstented subjects was 88.6% (109/123; 95% CI 81.6% to 93.6%), which exceeded the 50% performance goal (p<0.001), satisfying the primary efficacy objective. Primary patency by Kaplan-Meier analysis was 90.9% (95% CI 85.9% to 95.8%) through 12 months and 77.7% (95% CI 64.2% to 91.2%) through 390 days (Figure 2A). Freedom from CD-TLR by Kaplan-Meier analysis within 360 days was 97.1% (95% CI 94.3% to 99.9%; Figure 2B). The rate of CD-TLR was 2.9% (4/139; Table 3). Primary sustained clinical improvement was seen in 86.5% of subjects (115/133).
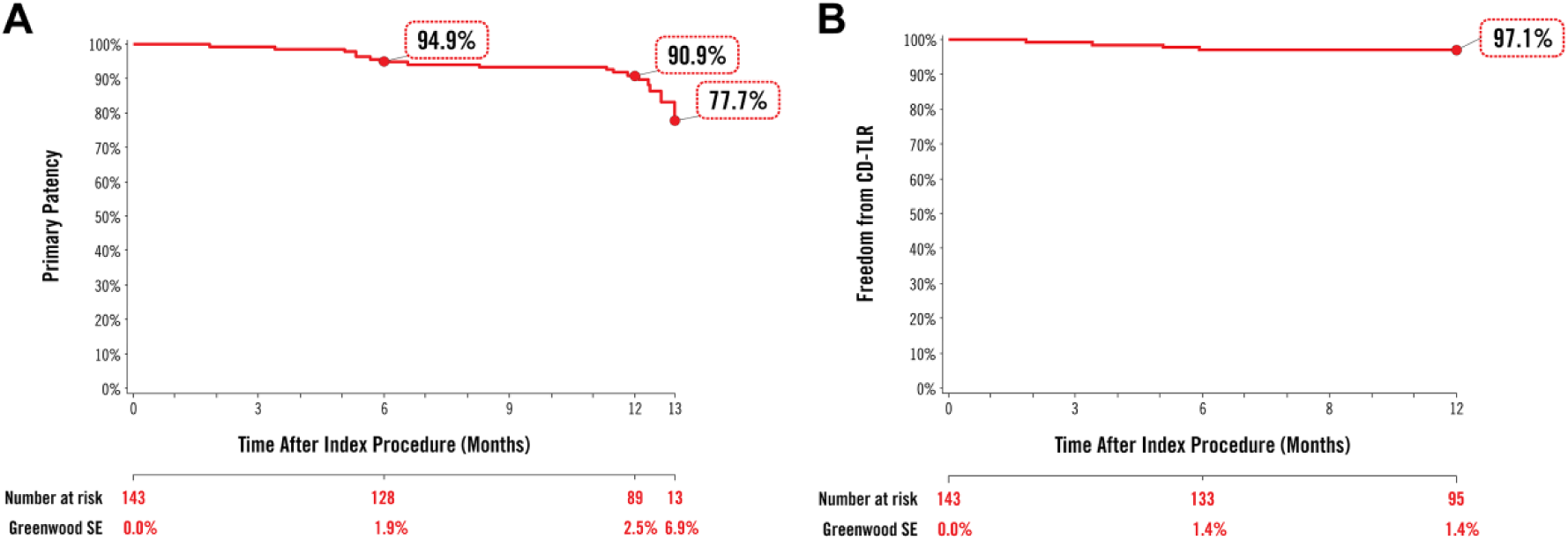
Kaplan-Meier estimates of (A) primary patency and (B) clinically-driven target lesion revascularization (CD-TLR) through 12 months. Numbers at clinical risk represent the evaluable subjects at the beginning of the 30-day window prior to each follow-up interval SE, standard error.
Safety and Effectiveness Outcomes at 12 Months. a
Abbreviations: TLR, target lesion revascularization; TVR, target vessel revascularization.
Data are given as the number/sample (percentage).
Primary patency is defined as freedom from clinically-driven TLR and freedom from restenosis as determined by duplex ultrasound peak systolic velocity ratio ≤2.4.
Any reintervention at the target lesion due to symptoms or drop of ankle-brachial index/toe-brachial index (ABI/TBI) of ≥20% or >0.15 when compared with postprocedure baseline ABI/TBI.
All TLR includes clinically-driven and incidental or duplex-driven TLR.
A sustained upward shift of at least 1 category on the Rutherford classification as compared to baseline without the need for repeated TLR or surgical revascularization in amputation-free survivors.
Freedom from device- and/or procedure-related mortality, freedom from major target limb amputation, and freedom from clinically-driven TLR within 30 days post-index procedure.
Composite of death, clinically-driven TVR, major target limb amputation, and thrombosis within 12 months.
Safety Outcomes
The primary safety objective of this trial was met. The primary safety composite outcome at 30 days was 99.3% (141/142, 95% CI 96.1% to 100.0%), compared to the performance goal of 88% (p<0.001). The MAE composite at 12 months was 4.3% (6/139 subjects). There were 4 CD-TVRs through 12 months, and all-cause death through 12 months was 2.9% (4/139). One death on day 7 from sudden cardiac death was adjudicated as procedure-related since it was in the first 30 days following the procedure. The other 3 subjects died due to gastrointestinal hemorrhage, sudden death, and an unknown cause, respectively. The rate of thrombosis was 2.2% (3/139). There were no amputations through 12 months.
Functional Outcomes
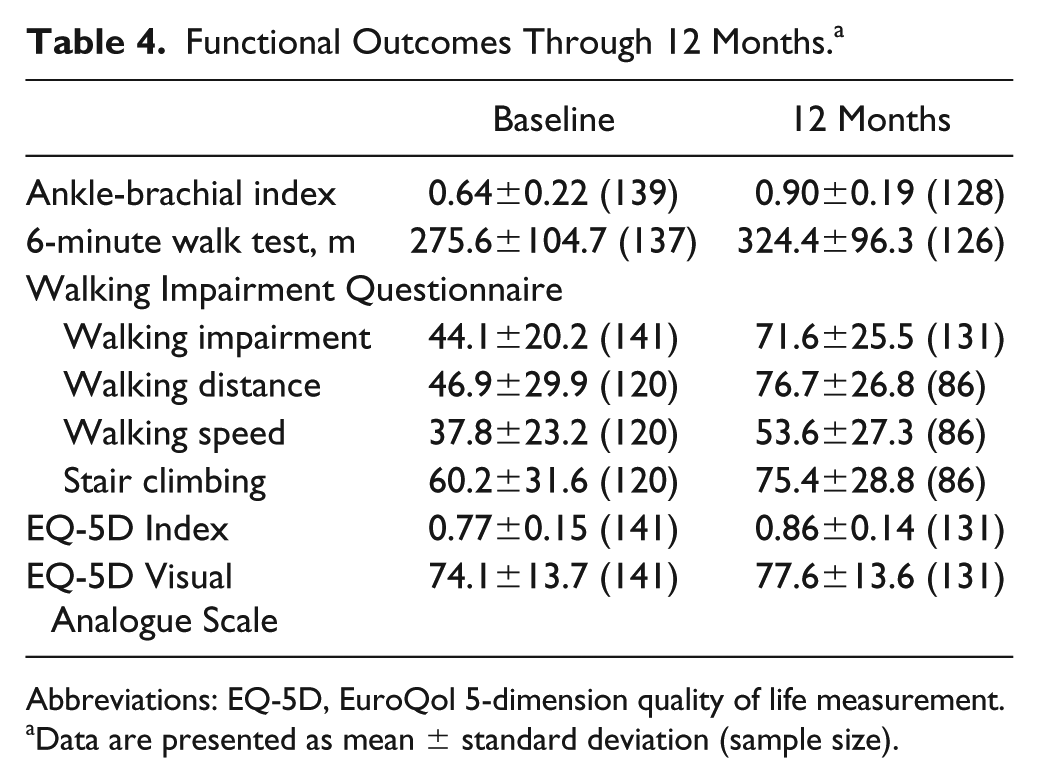
At 12 months, subjects showed improvement compared with baseline in all outcome metrics [Rutherford category, ankle-brachial index (ABI), WIQ, EQ-5D, and the 6MWT; Table 4]. Mean ABI at 12 months was 0.90±0.19, showing improvement from the baseline value of 0.64±0.22. For the 6-minute walk test, baseline values of 275.6±104.7 meters rose to 324.4±96.3 meters at 12-month follow-up, a substantial upward shift even in this complex subject cohort. Particularly notable was the transition of all CLI subjects (Rutherford category 4) to the claudication categories at the 12-month time point (Figure 3).
Functional Outcomes Through 12 Months. a
Abbreviations: EQ-5D, EuroQol 5-dimension quality of life measurement.
Data are presented as mean ± standard deviation (sample size).
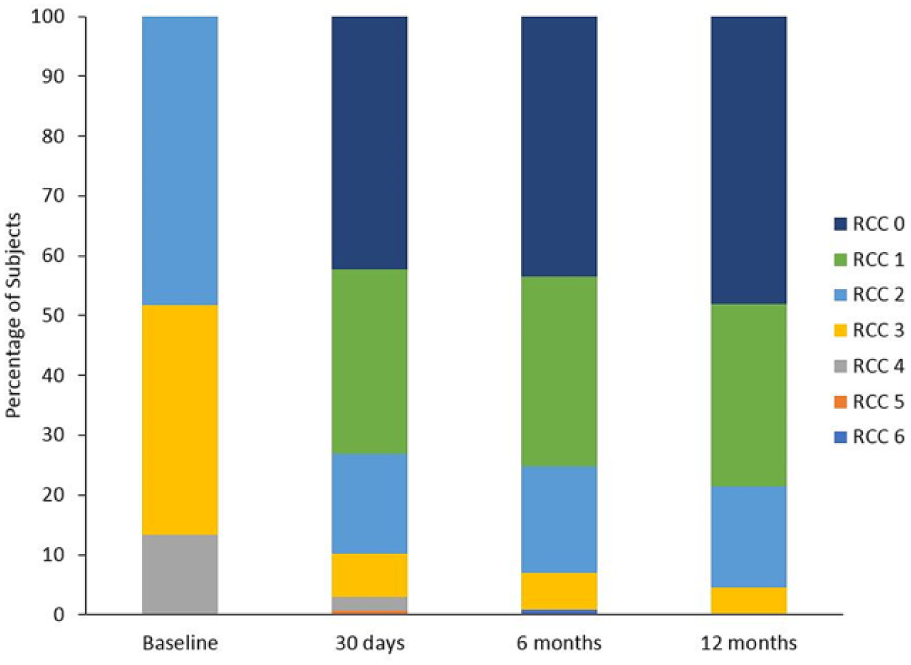
Change in Rutherford category through 12 months (p<0.001 between baseline and 12 months).
Discussion
The clinical evidence supporting the use of DCBs to treat femoropopliteal lesions has dramatically changed endovascular practice patterns in the United States and Europe. Randomized controlled trials and registries have shown that DCBs are safe and efficacious.8–18,34–36 However, the subjects in these trials were overwhelmingly Caucasian. As such, uncertainties remain as to the effectiveness of these and other medical devices to treat patients of other geographies and ethnic backgrounds. IN.PACT SFA China demonstrated high rates of patency and freedom from CD-TLR, providing additional data about the use of DCBs in China.
AcoArt I is the only other published RCT reporting on the use of DCBs in Chinese subjects. 15 AcoArt I involved 200 subjects recruited at 10 centers in China. It compared the use of Orchid, a paclitaxel-coated balloon that includes the carrier magnesium stearate, with an uncoated angioplasty balloon. Mean lesion length in this trial was 15.2±10.9 cm in the PTA arm and 14.7±11.0 cm in the DCB arm. The percentage of subjects in this trial treated with DCB classified with Rutherford category 5 disease was high (16%). At 12 months, primary patency by Kaplan-Meier analysis was 76.1% for DCB and 33.7% for PTA (p<0.001); the rate of TLR was 7.2% for DCB vs 39.6% for PTA (p<0.001). The provisional stenting rate was 19.0% for DCB and 21.0% for PTA (p=0.002), higher than the rate in this study. These outcomes clearly show the superiority of Orchid DCB compared with PTA.
Results from both trials support the emerging clinical rationale of using a metal implant only when necessary. In a progressive disease such as PAD, avoiding stent implantation can leave open a much larger number of treatment options for the interventionist if symptoms reoccur. However, many gaps remain when considering the data available for Chinese populations and the effect this has on the treatment algorithm. Currently, the standard of care for endovascular therapy in China is based on several factors: the lesion itself, device availability, the clinical presentation of the patient, and the economic position of the patient. As of late 2018, Acotec was the only DCB approved by China’s regulatory body; others were not available for use in China.
IN.PACT SFA China is the final 1-year dataset in the 4 studies comprising the IN.PACT DCB clinical program, and results continue to be consistent across trials. The 1-year patency estimate of 90.9% in IN.PACT SFA China was comparable to the rate of 87.5% from IN.PACT SFA, 37 as well as the rate of 93.9% in IN.PACT Japan. 17 Lesion lengths for the 3 DCB arms were similar: 10.4 cm in IN.PACT China, 8.9 cm in IN.PACT SFA, and 9.2 cm in IN.PACT Japan. Lesion and patient complexity are often difficult to consistently define hierarchically, and in these trials, different populations appear to have different characteristics that add to the overall difficulty in durably treating PAD. For example, the burden of diabetes was the highest in subjects enrolled in IN.PACT Japan (59.0%),17,18 with rates of 46.2% in IN.PACT China and 40.5% in IN.PACT SFA. 9 However, the level of concurrent below-the-knee disease was high (70.6%) in IN.PACT China, and the mean baseline ABI was lower (0.64) compared with IN.PACT SFA 9 (0.77) and IN.PACT Japan (0.76).17,18 Even so, the low CD-TLR rate of 2.9% through 1 year in IN.PACT China was also consistent with other IN.PACT DCB studies: 2.4% for IN.PACT SFA, 9 2.9% for IN.PACT Japan,17,18 and 3.8% for the post hoc ASEAN cohort 38 from IN.PACT Global. The 90.9% primary patency estimate by Kaplan-Meier analysis in this trial was comparable to other studies with 12-month follow-up, including AcoArt I, 15 LEVANT 2, 10 and ILLUMENATE,13,14 which reported patency estimates ranging from 73.5% to 89%.
Limitations
Limitations of this trial included a single-arm design with a limited number of subjects and follow-up through only 12 months. No economic data was analyzed in this study.
Conclusion
Results from IN.PACT SFA China demonstrated a high patency rate and few CD-TLRs at 12 months despite patient and lesion complexity. These results are consistent with other outcomes in the IN.PACT SFA DCB clinical program, showing the applicability of these results across a wide geography and patient population.
Supplemental Material
18-0408_Supplemental_tables – Supplemental material for IN.PACT SFA Clinical Study Using the IN.PACT Admiral Drug-Coated Balloon in a Chinese Patient Population
Supplemental material, 18-0408_Supplemental_tables for IN.PACT SFA Clinical Study Using the IN.PACT Admiral Drug-Coated Balloon in a Chinese Patient Population by Zhong Chen, Wei Guo, Weiliang Jiang, Feng Wang, Weiguo Fu, Yinghua Zou, Stefanie Deckers, Pei Li, Jeffrey J. Popma and Michael R. Jaff in Journal of Endovascular Therapy
Footnotes
Acknowledgements
The IN.PACT SFA China investigators would like to recognize and thank the patients involved with this clinical study for their participation. The authors acknowledge the following individuals from Medtronic for their contributions: Bridget Wall, PhD, for medical writing support and technical review; Eric Fernandez, MD, for technical review; and Anna Zhang and Kristy Sun for overall project management. Furthermore, the authors recognize all the principal investigators, subinvestigators, and institutions that enrolled in the study as collaborators in gathering this data (see ![]() ).
).
Authors’ Note
This study was presented at VEITH 2017 (November 13–17, 2017; New York City, NY, USA) and LINC 2018 (January 29–February 2, 2018; Leipzig, Germany).
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Wei Guo is the principal investigator of the AcoArt I study (AcoTec). Stefanie Deckers and Pei Li are full-time employees of Medtronic. Jeffrey J. Popma received institutional research grants from Medtronic, Boston Scientific, Abbott Vascular, and Cook and has received consulting income from Boston Scientific, Cordis, and Edwards Lifesciences. Michael R. Jaff is a noncompensated advisor for Medtronic. He has an equity investment in PQ Bypass and is a paid consultant to Philips/Volcano and Vactronix.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Medtronic.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.