
Abstract
Keywords
Introduction
Atherosclerotic peripheral artery disease (PAD) involving the superficial femoral artery (SFA) and/or popliteal arteries plays a central role in lifestyle-limiting claudication and critical limb ischemia (CLI). Drug-coated balloons (DCBs) were developed to overcome the challenges of traditional implantable drug-eluting and non-drug-based endovascular interventions. Currently, DCBs are coated with the antiproliferative agent paclitaxel, which transfers to the inner arterial wall on inflation and may persist in the vessel tissue for up to 180 days.1–3
Numerous randomized controlled trials (RCTs) have demonstrated that DCBs are safe and more effective for the treatment of patients with femoropopliteal lesions than uncoated balloon angioplasty (IN.PACT SFA, LEVANT 2, BIOLUX P-1, THUNDER, ILLUMENATE EU, ILLUMENATE Pivotal).4–10 Meta-analyses of clinical trials have also shown that DCBs are a cost-effective option that provides clinically meaningful benefit compared with traditional non-drug-based treatments, such as bare metal stents or angioplasty with an uncoated balloon.11,12 However, randomized investigational device exemption (IDE) trials in the United States are characterized by strict enrollment criteria guided by discussions with the Food and Drug Administration (FDA) to ensure uniform patient populations. These criteria typically exclude complex but frequently encountered pathologies, such as long lesions [typically described as TransAtlantic Inter-Society Consensus II (TASC) C and D], de novo in-stent restenosis (ISR), long chronic total occlusions (CTO), and severe calcification in the most common classification schemes.
RCTs conducted elsewhere (THUNDER, FEMPAC, PACIFIER, CONSEQUENT, AcoArt I) typically include more complex patient cohorts but also fewer patients.13–17 Different from trials in the United States, they report a primary outcome of late lumen loss using core laboratory–adjudicated angiography at 6 months and site-reported target lesion revascularization (TLR). Importantly, all demonstrated the clinical benefit of treatment with a DCB. However, there continues to be a need for larger controlled and adjudicated datasets on the use of DCBs in patients with complex lesions that are seen in everyday practice.
The IN.PACT Global Study is a prospective, independently adjudicated trial that evaluated the use of DCBs for patients with a broad range of lesion types in the native SFA and/or popliteal artery. 18 The study design included 3 prespecified cohorts for the prospective analysis of DCB safety and effectiveness in patients with distinct complex lesion types: long lesions ⩾15 cm, de novo ISR, and CTO ⩾5 cm. Twelve-month results from each of these analyses have been reported and support the safety and effectiveness of DCB use for each of these complex lesion types.19–21
This report details a post hoc analysis comparing 12-month clinical outcomes between subjects in the IN.PACT Global Study clinical cohort with clinical and lesion characteristics that are routinely included in RCTs (standard-use group) vs those typically excluded from RCTs owing to increased complexity (broader-use group).
Methods
Study Design
The IN.PACT Global Study is a prospective, multicenter, international, single-arm clinical study assessing the safety and effectiveness of the paclitaxel-coated IN.PACT Admiral DCB (Medtronic Inc., Dublin, Ireland) for the treatment of real-world patients with intermittent claudication and/or ischemic rest pain due to atherosclerotic disease of the femoropopliteal segment, including the entire native SFA and/or popliteal artery. The trial was registered on the National Institutes of Health website (ClinicalTrials.gov; identifier NCT01609296). The institutional review board or ethics committee at each study site approved the study protocol. Informed consent was obtained from all subjects before enrollment. The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and applicable laws as specified by all relevant governmental bodies. An independent Clinical Events Committee (CEC; Syntactx, New York, NY, USA) composed of interventional and noninvasive clinicians was established to assess the primary and certain secondary endpoints and to determine whether each met protocol-specified criteria.
The IN.PACT Global Study enrolled subjects with symptoms of intermittent claudication and/or ischemic rest pain (Rutherford categories 2–4) and angiographic evidence of ⩾2-cm long stenosis or occlusion [de novo or restenosis (in-stent or native vessel)] in the SFA and/or popliteal artery (including P1–P3 segments). Details of the DCB device, study design, and treatment parameters have been previously described. 18 Pre- and postdilation were permitted at the discretion of the investigator. Vessel preparation by other devices was not permitted, and postdilation could be performed using an uncoated balloon only.
The current post hoc analysis compared 12-month clinical outcomes between subjects who would have been included in the IN.PACT SFA RCT vs those patients who would have been excluded. The flow of patients is shown in Figure 1. Of the 1535 subjects enrolled in IN.PACT Global, 119 were included in the 150-mm DCB cohort and used for regulatory purposes. These subjects were not included in the full cohort as they were not consecutively enrolled, introducing a potential treatment bias. In addition, 10 patients were not treated with a DCB, resulting in a full clinical cohort of 1406 subjects treated with a DCB (intention-to-treat population). The 1- and 2-year results of this cohort have been presented, and the imaging cohorts have been presented (CTO, long lesion)20,21 and published (ISR). 19
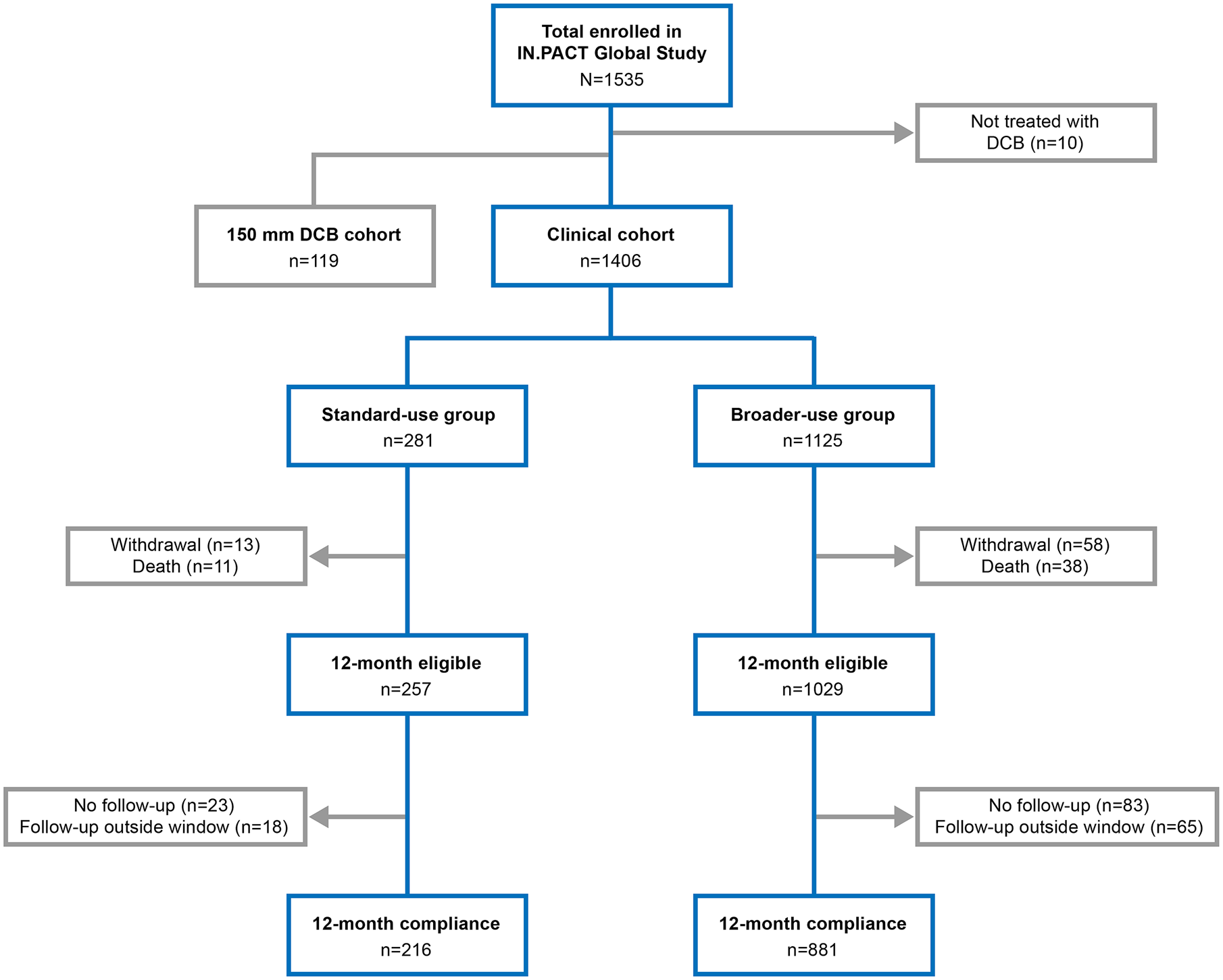
Patient flow in the standard-use and broader-use groups in the IN.PACT Global Study clinical cohort through 12 months. DCB, drug-coated balloon.
For this analysis, it was necessary to create a cohort typically enrolled in an RCT, so subjects from the 1406 clinical cohort patients were retrospectively assigned to a standard-use group based on the inclusion/exclusion criteria employed in the randomized IN.PACT SFA trial. 9 The 281 assigned patients had single 70% to 99% stenotic de novo or native artery restenotic lesions ⩽18 cm long or CTOs ⩽10 cm long. Lesions could have mild calcification but ISR was not allowed. The remaining 1125 clinical cohort patients were assigned to a broader-use group that included subjects who would have been excluded from the trial, namely, those with multiple lesions, single de novo lesions >18 cm, CTOs >10 cm, any ISR, moderate or severe calcification, or bilateral disease. Calcification was categorized as mild (on one side of the artery only and over less than half the total lesion length), moderate (one side of the artery only and over greater than or equal to half the total lesion length), moderately severe (both sides of the artery at the same location over less than half the total lesion length), and severe (both sides of the artery at the same location over greater than or equal to half the total lesion length). 22
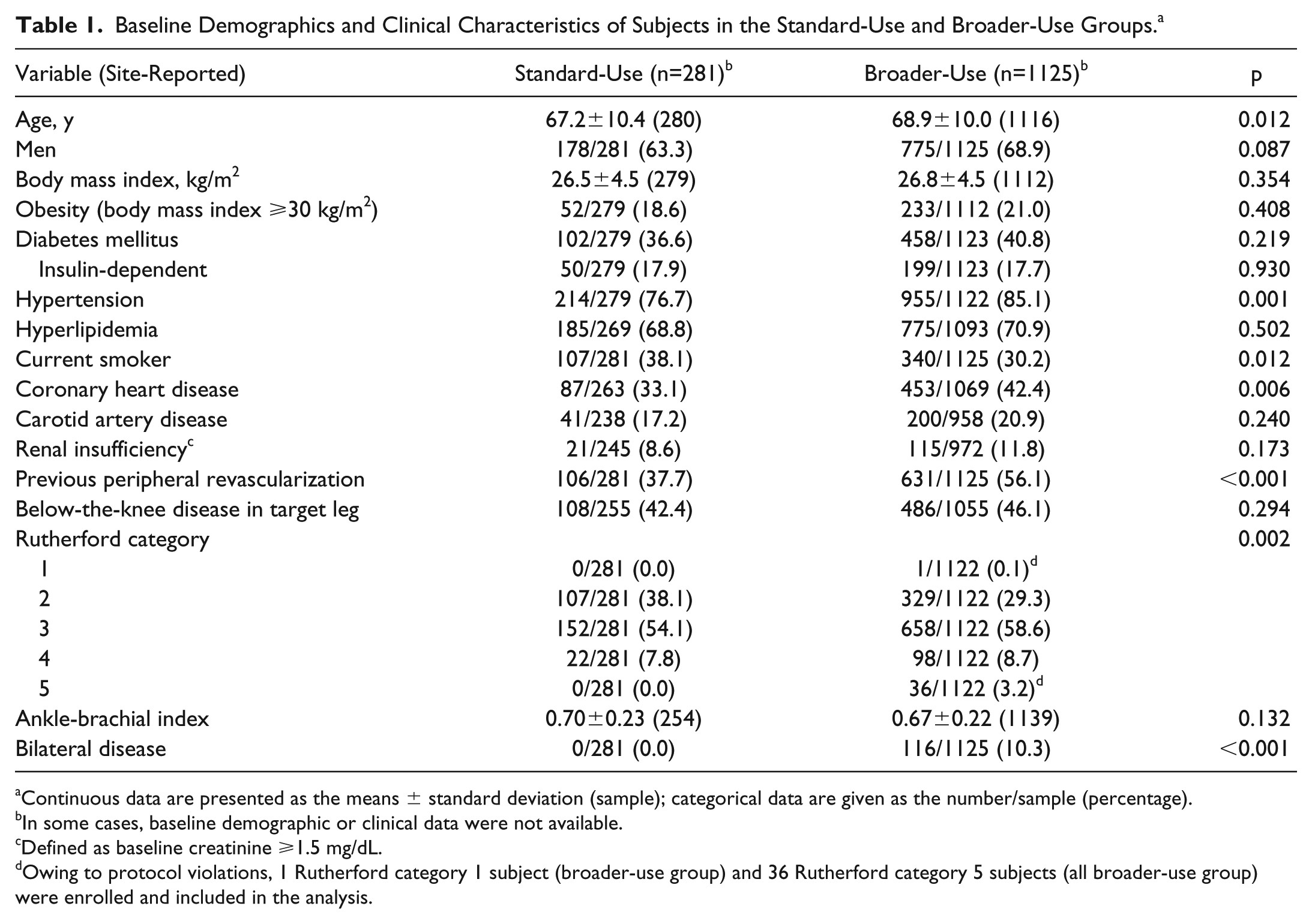
Site-reported baseline demographics and lesion characteristics are reported in Tables 1 and 2. Nineteen of the 116 subjects with bilateral disease had only standard lesions but were classified in the broader-use group because both legs were affected (1.7% of the 1125 patients).
Baseline Demographics and Clinical Characteristics of Subjects in the Standard-Use and Broader-Use Groups. a
Continuous data are presented as the means ± standard deviation (sample); categorical data are given as the number/sample (percentage).
In some cases, baseline demographic or clinical data were not available.
Defined as baseline creatinine ⩾1.5 mg/dL.
Owing to protocol violations, 1 Rutherford category 1 subject (broader-use group) and 36 Rutherford category 5 subjects (all broader-use group) were enrolled and included in the analysis.
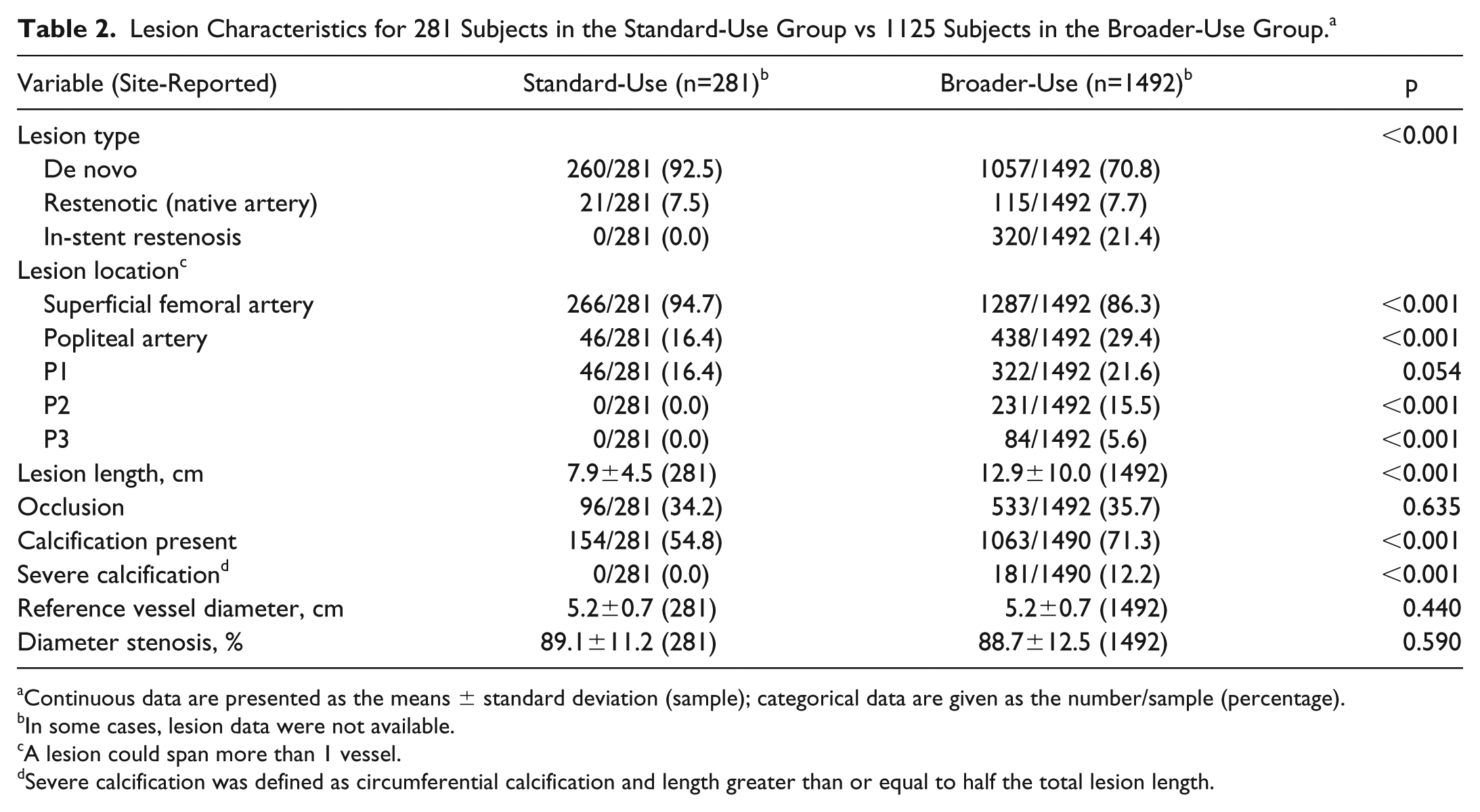
Lesion Characteristics for 281 Subjects in the Standard-Use Group vs 1125 Subjects in the Broader-Use Group. a
Continuous data are presented as the means ± standard deviation (sample); categorical data are given as the number/sample (percentage).
In some cases, lesion data were not available.
A lesion could span more than 1 vessel.
Severe calcification was defined as circumferential calcification and length greater than or equal to half the total lesion length.
Study Endpoints
The composite primary safety endpoint was freedom from device- and procedure-related mortality through 30 days and freedom from 12-month major target limb amputation and clinically-driven target vessel revascularization (CD-TVR). The latter was assessed at the subject level and defined as the first event that required revascularization in the subject. The primary effectiveness endpoint was clinically-driven target lesion revascularization (CD-TLR) within 12 months. The CEC reviewed all CD-TLR and CD-TVR events to determine which were clinically driven, that is, due to symptoms or an ankle-brachial index (ABI) decrease ⩾20% or >0.15 when compared with the baseline value. Clinically-driven TLRs or TVRs did not include procedures that were performed on asymptomatic subjects or that were based only on diagnostic imaging procedures.
Secondary endpoints included CD-TLR, the incidence of major adverse events (all-cause mortality, CD-TVR, major target limb amputation, thrombosis at the target lesion site), any TLR, and any TVR at 12 months. Other secondary outcomes were the change in Rutherford category and primary sustained clinical improvement (defined as freedom from major target limb amputation, freedom from TVR, and improvement in Rutherford category). The CEC adjudicated all major adverse events.
Functional assessments included the ABI, EuroQol 5-Dimension Quality of Life Index (EQ-5D), the Walking Impairment Questionnaire (WIQ), and the nights in hospital (a cumulative measure over the 12-month follow-up that included any time a subject returned to the hospital for any issue related to the study lesion). Acute periprocedural outcomes (device success, procedure success, and clinical success) and categories of provisional stenting (spot stenting and partial lesion coverage) were previously defined. 18
Statistical Analysis
All data used in the analysis were provided by the investigational sites and all summaries were based on nonmissing assessments. Unless otherwise specified, all baseline demographics and clinical characteristics were summarized on a subject basis; lesion characteristics were summarized on a lesion basis. For baseline characteristics, continuous variables were described as mean ± standard deviation and were compared using a 2-sample t test. Dichotomous and categorical variables were described as numbers (proportions) and were compared with the Fisher exact test or Cochran-Mantel-Haenszel modified ridit scores, respectively. For functional outcomes, change from baseline within each group was compared with Wilcoxon signed rank test, and differences between groups were compared with the Wilcoxon rank sum test.
The Kaplan-Meier method was used to evaluate time-to-event data for per-patient freedom from CD-TLR over the 12-month follow-up; the log-rank test was used for the group comparisons. For event rates that were expressed as a proportion, the number of subjects with an event within 360 days was the numerator and the total number of subjects with an event or at least 300 days of clinical follow-up was the denominator. Assessment of clinical characteristics from baseline to 12 months involved subjects who had data at both time points. Statistical analyses were performed using SAS (version 9.4; SAS Institute, Cary, NC, USA)
Results
Group Comparison
Compared with the standard-use (SU) patients, the broader-use (BU) patients were older (p=0.012) and had more hypertension (p=0.001), coronary heart disease (p=0.006), previous peripheral interventions (p<0.001), and more severe ischemia (Rutherford categories 3–5; p=0.002) but fewer current smokers (p=0.012; Table 1). The BU group also had longer lesions (p<0.001), a higher percentage of calcified lesions (p<0.001), more popliteal involvement (p<0.001; Table 2) and included patients with bilateral disease (Table 1) and ISR (excluded from the SU group; Table 2).
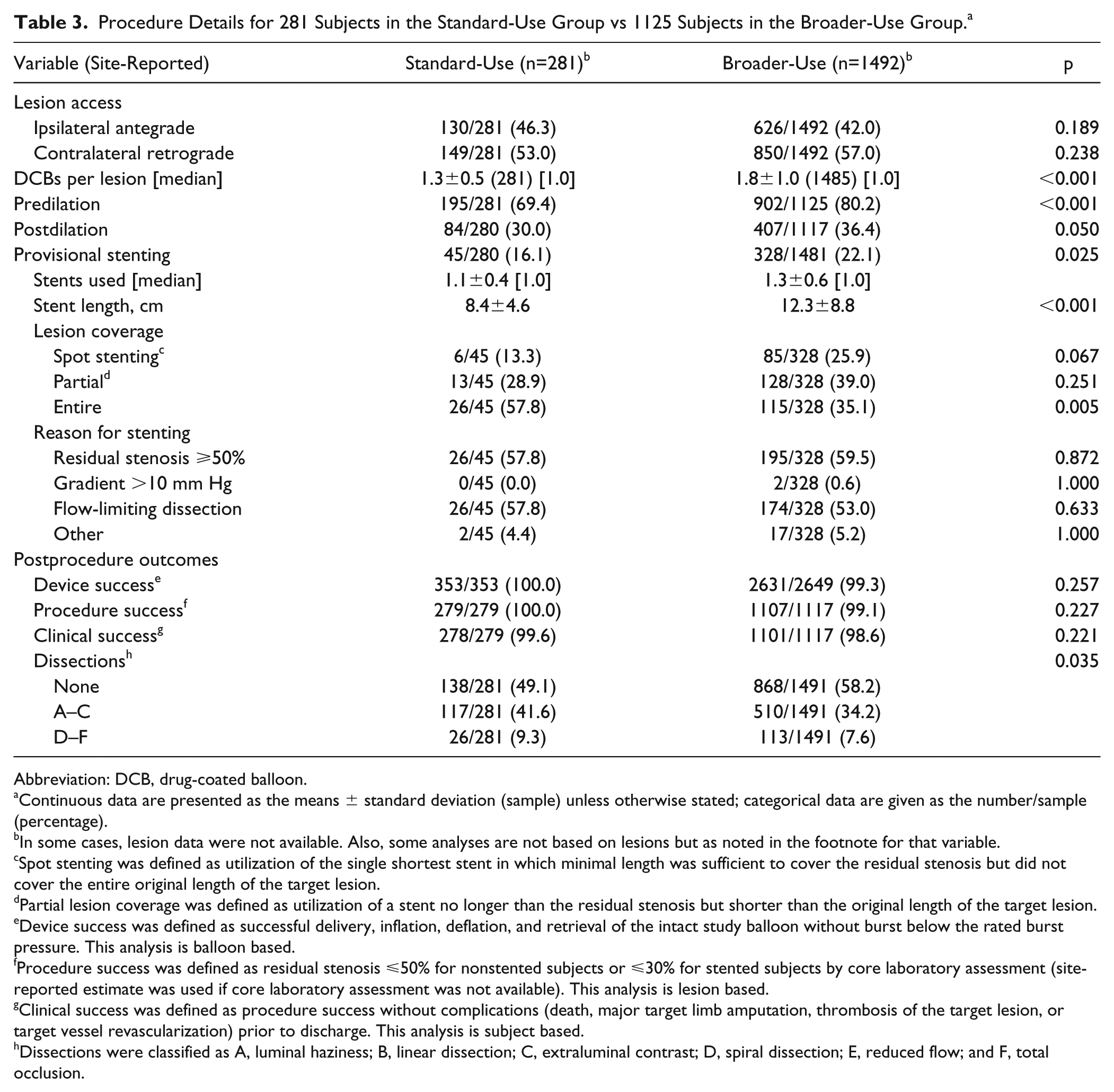
Predilation was performed more frequently in subjects from the BU group (902/1125, 80.2%) compared with the SU group (195/281, 69.4%; p<0.001). Postdilation was performed in 36.4% (407/1117) of subjects in the BU group vs 30.0% (84/280) of SU patients (p=0.050; Table 3). Provisional stents were implanted more frequently in the BU group (328/1481, 22.1%) compared with the SU patients (45/280, 16.1%; p=0.025). The mean stent length was also longer in the BU (12.3±8.8 cm) compared with the SU patients (8.4±4.6 cm, p<0.001). Lesion coverage and reasons for stenting are reported in Table 3.
Procedure Details for 281 Subjects in the Standard-Use Group vs 1125 Subjects in the Broader-Use Group. a
Abbreviation: DCB, drug-coated balloon.
Continuous data are presented as the means ± standard deviation (sample) unless otherwise stated; categorical data are given as the number/sample (percentage).
In some cases, lesion data were not available. Also, some analyses are not based on lesions but as noted in the footnote for that variable.
Spot stenting was defined as utilization of the single shortest stent in which minimal length was sufficient to cover the residual stenosis but did not cover the entire original length of the target lesion.
Partial lesion coverage was defined as utilization of a stent no longer than the residual stenosis but shorter than the original length of the target lesion.
Device success was defined as successful delivery, inflation, deflation, and retrieval of the intact study balloon without burst below the rated burst pressure. This analysis is balloon based.
Procedure success was defined as residual stenosis ⩽50% for nonstented subjects or ⩽30% for stented subjects by core laboratory assessment (site-reported estimate was used if core laboratory assessment was not available). This analysis is lesion based.
Clinical success was defined as procedure success without complications (death, major target limb amputation, thrombosis of the target lesion, or target vessel revascularization) prior to discharge. This analysis is subject based.
Dissections were classified as A, luminal haziness; B, linear dissection; C, extraluminal contrast; D, spiral dissection; E, reduced flow; and F, total occlusion.
Safety Outcomes
The 12-month primary safety composite endpoint was met by proportionally fewer subjects in the BU group [91.0% (952/1046)] than in the SU cohort [96.2% (255/265), p=0.003] and was mainly driven by a higher incidence of CD-TVR in the BU patients [9.1% (95/1046)] vs the SU patients [4.2% (11/265), p=0.008; Table 4]. The rate of all-cause death at 12 months was not significantly different between groups [3.4% (36/1046) BU vs 3.8% (10/265) SU; p=0.852]. Independent adjudication by the CEC determined that none of the deaths was related to the study device. Two of the deaths in the BU group and 1 in the SU group occurred within 30 days of the index procedure, so the CEC adjudicated them as possibly or potentially procedure-related. Details of these events have been previously reported. 18
12-Month Safety Outcomes in the Standard-Use and Broader-Use Groups. a
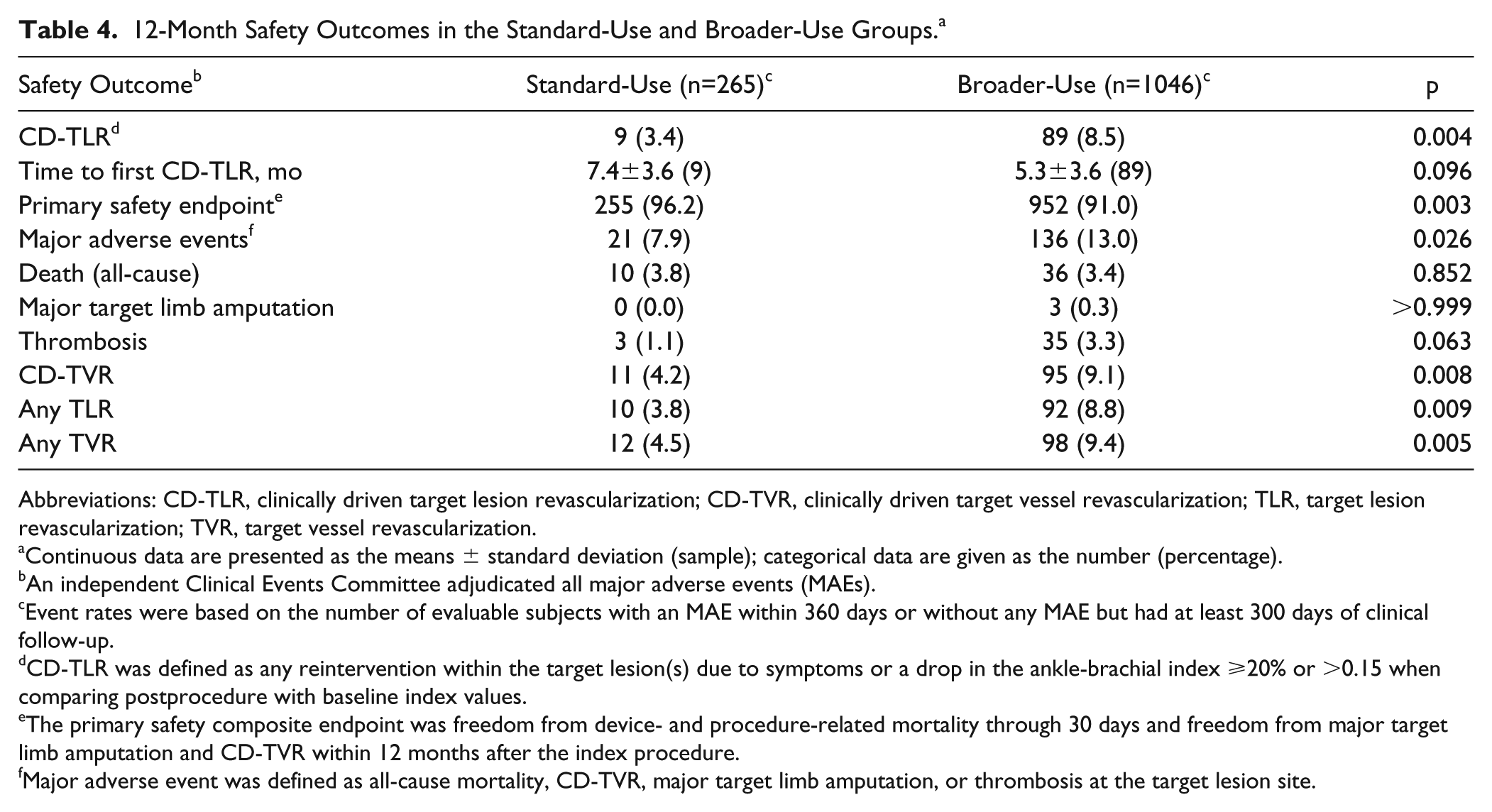
Abbreviations: CD-TLR, clinically driven target lesion revascularization; CD-TVR, clinically driven target vessel revascularization; TLR, target lesion revascularization; TVR, target vessel revascularization.
Continuous data are presented as the means ± standard deviation (sample); categorical data are given as the number (percentage).
An independent Clinical Events Committee adjudicated all major adverse events (MAEs).
Event rates were based on the number of evaluable subjects with an MAE within 360 days or without any MAE but had at least 300 days of clinical follow-up.
CD-TLR was defined as any reintervention within the target lesion(s) due to symptoms or a drop in the ankle-brachial index ⩾20% or >0.15 when comparing postprocedure with baseline index values.
The primary safety composite endpoint was freedom from device- and procedure-related mortality through 30 days and freedom from major target limb amputation and CD-TVR within 12 months after the index procedure.
Major adverse event was defined as all-cause mortality, CD-TVR, major target limb amputation, or thrombosis at the target lesion site.
There were no statistically significant differences between groups in the rates of major target limb amputation (p>0.999) or thrombosis (p=0.063; Table 4). Though the rate of thrombosis was low in both groups, there was a slight trend to more thrombosis in the BU group, which may not be unexpected given the complexity of the disease.
Effectiveness Outcomes
The Kaplan-Meier estimate of freedom from CD-TLR at 12 months was 91.6% in the BU group vs 96.6% in the SU patients (log-rank p=0.005; Figure 2). Freedom from CD-TLR at 13 months was 89.7% vs 95.4%, respectively (log-rank p=0.004). Twelve-month CD-TLR occurred more frequently in the BU group [8.5% (89/1046)] compared with the SU group [3.4% (9/265), p=0.004]. There was no difference in mean time to first CD-TLR between groups (5.3±3.6 months BU vs 7.4±3.6 months SU, p=0.096; Table 4).
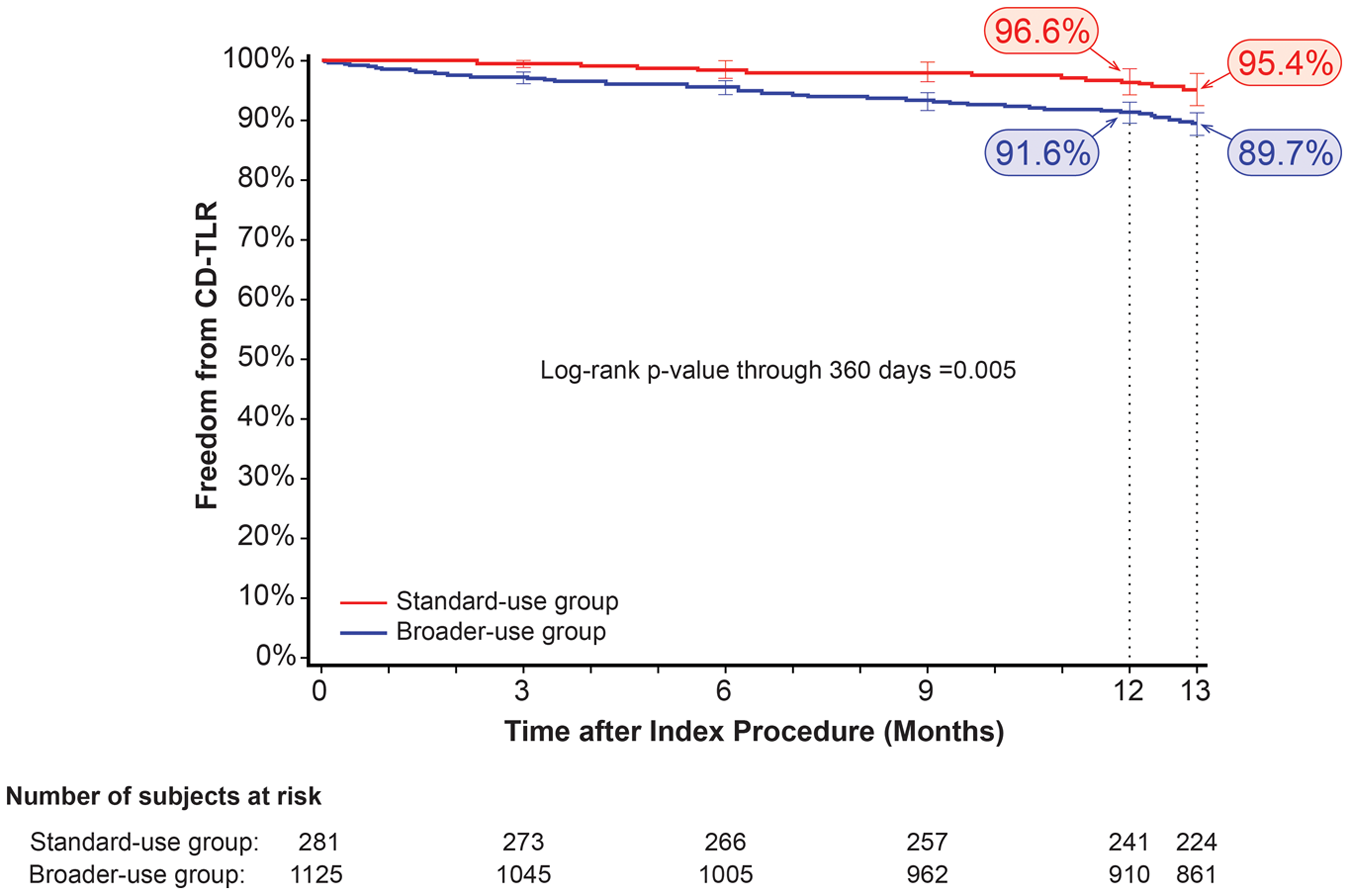
Kaplan-Meier curve of freedom from clinically driven target lesion revascularization (CD-TLR) through 12 months in the standard-use and broader-use groups. Numbers at risk represent the patients at the beginning of the 30-day window prior to each follow-up interval.
Functional Outcomes
Both groups had significant improvements in Rutherford class from baseline to 12 months (p<0.001; Table 5). There was no significant difference between the changes in each group (p=0.087). The rate of primary sustained clinical improvement was lower in the BU group [78.7% (747/949) vs the SU patients [88.0% (206/234), p=0.001] at 12 months (Table 6). Mean EQ-5D scores were significantly improved from baseline to 12 months in both groups (p<0.001). Mean change from baseline in EQ-5D score at 12 months was significantly higher for the SU group compared with the BU patients (p<0.001; Table 6).
Changes in Rutherford Category at 12 Months in the Standard-Use and Broader-Use Groups. a
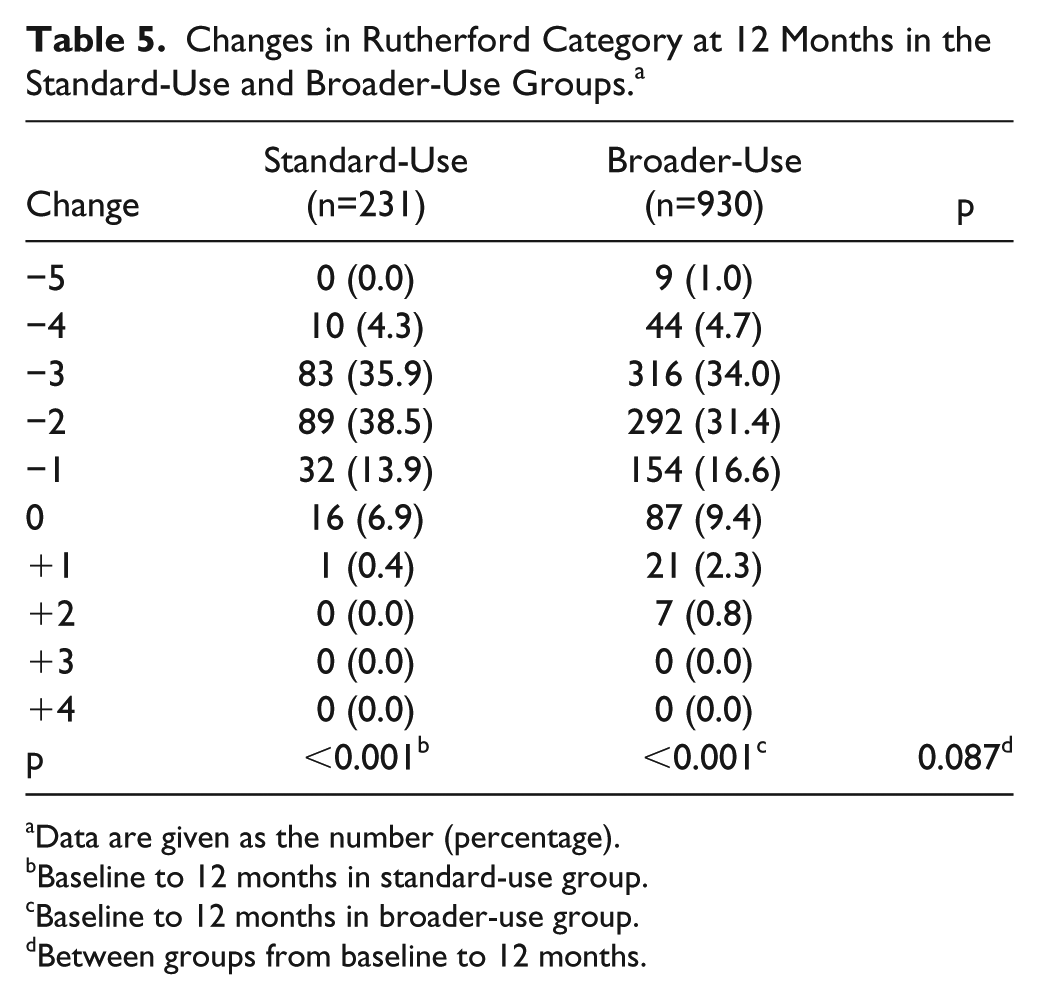
Data are given as the number (percentage).
Baseline to 12 months in standard-use group.
Baseline to 12 months in broader-use group.
Between groups from baseline to 12 months.
Twelve-Month Functional Outcomes in the Standard-Use and Broader-Use Groups. a
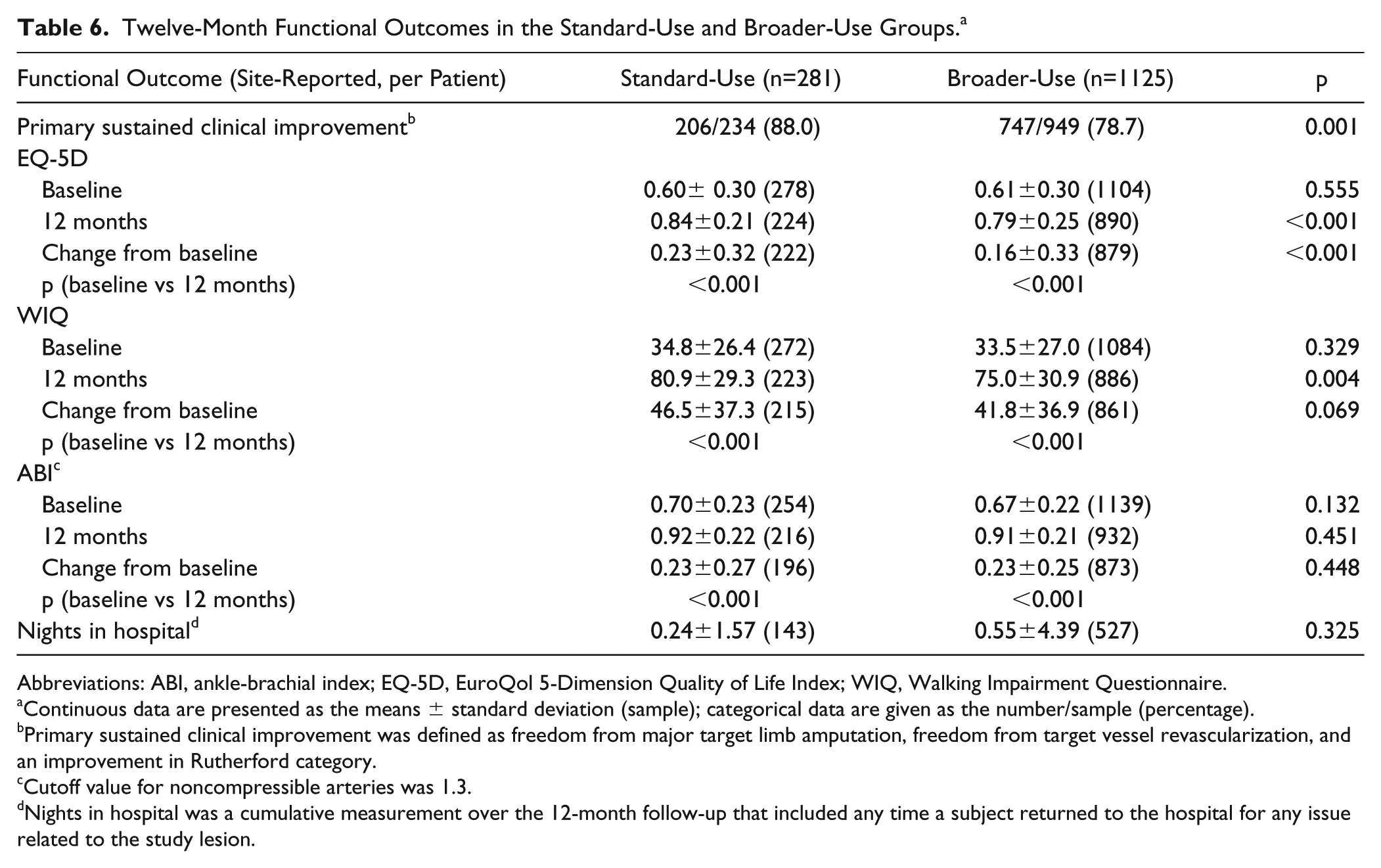
Abbreviations: ABI, ankle-brachial index; EQ-5D, EuroQol 5-Dimension Quality of Life Index; WIQ, Walking Impairment Questionnaire.
Continuous data are presented as the means ± standard deviation (sample); categorical data are given as the number/sample (percentage).
Primary sustained clinical improvement was defined as freedom from major target limb amputation, freedom from target vessel revascularization, and an improvement in Rutherford category.
Cutoff value for noncompressible arteries was 1.3.
Nights in hospital was a cumulative measurement over the 12-month follow-up that included any time a subject returned to the hospital for any issue related to the study lesion.
Mean WIQ scores and ABI were also significantly improved from baseline to 12 months in both groups (all p<0.001). For both measures, the mean change from baseline to 12 months was similar between groups (WIQ, p=0.069; ABI, p=0.448). There was no difference in the median ABI at baseline between groups, whether the cutoff of 1.3 or 1.4 was used to define noncompressible arteries. Mean nights in hospital were 0.55±4.39 for the BU group and 0.24±1.57 for the SU patients (p=0.325; Table 6).
Discussion
The IN.PACT Global Study evaluated real-world patients with adjudication of subgroups based on complex, often difficult to treat lesion types, including long lesions (⩾15 cm), de novo ISR, and CTOs (⩾5 cm); the effectiveness of DCB treatment was reflected in the 92.6% freedom from CD-TLR in a heterogeneous population. 18
The current post hoc analysis was performed to compare 12-month outcomes between subjects with clinical and lesion characteristics that are usually included in RCTs and those that are typically excluded but commonly seen in daily practice. The results demonstrate DCB performance that is parallel with not only strictly controlled RCTs but also trials of DCBs that have been specifically designed to evaluate performance in patients with complex femoropopliteal lesions. For example, the 96.6% estimated freedom from CD-TLR in the SU group was similar to the 97.5% estimate from the IN.PACT SFA RCT of TASC II A/B lesions. 9 Similarly, the 91.6% estimate for CD-TLR in the BU group (22% stented) was comparable to data for different types of complex lesions from the IN.PACT Global imaging cohorts (94.2% for long lesions with 40.4% stenting, 20 92.9% for de novo ISR with 14.5% stenting, 19 and 89.1% for CTO with 46.8% stenting 21 ).
The IN.PACT Global Study was designed to enroll a real-world patient population, and when subjects in the full clinical cohort were retrospectively dichotomized based on the selection criteria used in the IN.PACT SFA RCT, only 20% of subjects were assigned to the SU group. There are multiple implications to this observation. First, strict enrollment criteria for randomized IDE trials mandated by the FDA severely limit the number of patients who will be eligible for participation in an RCT. Perhaps more importantly, the results of an RCT may be generalizable only to the small fraction of patients meeting the enrollment criteria. Notably, those RCTs conducted outside the United States have different enrollment criteria, often allowing more severe patients to participate. This is commendable and allows for a more diverse patient type to be studied in a randomized fashion. However, the number of patients in these trials is usually smaller, and endpoints are not uniformly reported.
Second, it is necessary to adopt a comprehensive approach that includes more than RCTs, including post hoc analyses and global registries, to provide evidence of safety and effectiveness in the other 80% of patients who would typically be excluded from an RCT. Finally, 12-month clinical outcomes from the RCT-simulated SU group (96.6% freedom from CD-TLR) were parallel to data from the IN.PACT SFA RCT of the Admiral DCB (97.5% freedom from CD-TLR), substantiating the current post hoc approach for evaluating treatments in SU vs BU groups. 9
Bilateral disease, regardless of lesion type, was a criterion for automatic assignment to the BU group; 1.7% of subjects in the BU group had lesions that would have assigned them to the SU group, but because lesions were present in both legs, the subjects were assigned to BU. Although these lesions are more easily treated and could be argued to have increased the rates of success in the BU group, this small fraction of patients was not likely to have a significant impact on the results. Nearly 9% of the bilateral disease subjects in the BU group had at least 1 lesion that would have disqualified them from assignment to the SU group.
DCB treatment appears safe among patients in the BU group. Although the safety composite endpoint was met by a significantly smaller percentage of BU patients (driven by a higher rate of CD-TVR), the endpoint was still met by >90% of patients in the BU group. Of significance for the practicing clinician, there were no differences in the incidence of most individual major adverse events (all-cause death, major target limb amputation, and thrombosis).
Treatment with a DCB led to clinically meaningful improvements in all functional and quality-of-life measures between baseline and 12 months in each group. The difference in outcomes between the groups was the result of a higher TVR rate in the BU patients. Improved function and quality of life in SU patients is consistent with RCTs that have demonstrated the clinical value and cost-effectiveness of DCBs in patients with femoropopliteal disease.11,12,23 The added value of the current analysis is that DCB treatment is now associated with functional and quality-of-life improvements in an additional population of patients typically excluded from RCTs but common in everyday practice.
The decrease in quality of life from claudication is severe and of similar magnitude to that of other significant chronic diseases. 24 Functional and quality-of-life outcomes are used across multiple therapeutic areas to evaluate the clinical value of various treatment options, often in the larger context of cost effectiveness. Though few question the clinical benefit of total knee arthroplasty procedures for patients with osteoarthritis of the knee, a recent analysis revealed that knee replacement surgery had minimal effect on overall quality of life or cost savings in an overall cohort of patients spanning a range of arthritis severity. 25 A deeper analysis revealed that quality of life and cost savings could improve if knee replacement surgery were limited to patients with severe knee osteoarthritis. 25 In this way, the current IN.PACT Global Study post hoc analysis demonstrated that treatment with a DCB is safe and effective and leads to clinically meaningful improvements not only in patients with characteristics that are typical of RCTs but also in those with more complex disease usually excluded from RCTs.
Limitations
As a single-arm study that includes all-comers, this study does not have a control or active comparator group, unlike RCTs. As such, the results do not allow direct comparison to other DCBs or endovascular modalities. In addition, the prospective evaluation of anatomic outcomes by duplex ultrasonography and/or angiography was limited to subjects who were assigned to predefined cohorts (long lesion, de novo ISR, and CTO), which means that data from the current post hoc analysis were obtained from the investigational sites. Thus, analyses that were performed on the entire clinical cohort (imaging and nonimaging components) were limited to clinical outcomes, and an independent CEC reviewed and adjudicated all events that were reported as part of these outcomes (CD-TLR, CD-TVR, and major adverse events).
Conclusion
Patients with PAD present with a broad range of clinical and lesion characteristics, but RCTs, especially those in the US, typically exclude most patients with complex lesions that are commonly seen in everyday practice. This post hoc analysis demonstrated that the paclitaxel IN.PACT Admiral DCB appears safe, with clinically important effectiveness at 12 months in the treatment of patients with lesions that are typically excluded from an RCT, although there were increased incidences of CD-TLR and CD-TVR. Moving forward, there is a need for a comprehensive approach that includes RCTs, post hoc analyses, and global registries that can provide information about the safety and effectiveness of endovascular treatment options in patients with lesions of any type or complexity.
Footnotes
Acknowledgements
We would like to recognize and thank the patients involved with this clinical study for their participation. The authors thank Bridget Wall, PhD, for technical review of the manuscript and Zachary Harrelson, PhD, of Meridius Health Communications Inc, San Diego, CA, for providing medical writing support, which was funded by Medtronic Inc, Minneapolis, MN, in accordance with Good Publication Practice (GPP3) guidelines (![]() ).
).
Authors’ Note
Preliminary study data were presented at the Charing Cross Symposium (April 25, 2017; London, UK).
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Gary Ansel is an advisor for Abbott Vascular, Boston Scientific Corp., Medtronic, Philips & Volcano Philips, and W.L. Gore & Associates; a compensated board member of VIVA Physicians, a 501c3 not-for-profit education and research organization; an equity investor in Embolitech and Primacea; and the recipient of royalties from Cook Medical. Marianne Brodmann has received speaking honoraria from Bard Peripheral Vascular, Biotronik, Medtronic, Spectranetics, and Viva Physicians and is a consultant for Bard Peripheral Vascular, Biotronik, Medtronic, and Spectranetics. Antonio Micari is a compensated consultant for Medtronic and Boston Scientific Corp. Michael Jaff has served as a non-compensated advisor for Medtronic; is an equity investor of PQ Bypass, Embolitech, and Vascular Therapies; and is an advisor to Philips/Volcano and Vactronix. Krishna Rocha-Singh is a compensated consultant for Medtronic, Zimmer-BioMet, ROX Medical, Alucent Medical, SoundBite Medical, Daiichi Sankyo, and Abbott Vascular; a compensated board member of VIVA Physicians, a 501c3 not-for-profit education and research organization; and an equity investor in PQ Bypass. Eric Fernandez and Hong Wang are full-time employees of Medtronic. Thomas Zeller is a consultant for Abbott Vascular, Bard Peripheral Vascular, Boston Scientific Corp., Cook Medical, W.L. Gore & Associates, GLG, Medtronic, and Spectranetics and has received speaking honoraria from Abbott Vascular, Bard Peripheral Vascular, Biotronik, Boston Scientific Corp., Cook Medical, W.L. Gore & Associates, Medtronic, Philips, Spectranetics, Straub Medical, TriReme, Veryan, and VIVA Physicians. His clinic has received study funds or funds for research or clinical trials from 480 Biomedical, Abbott Vascular, B. Braun, Bard Peripheral Vascular, Bayer Pharma, Biotronik, Caveo Med, Contego Medical, Cook Medical, CSI, W.L. Gore & Associates, Innora, Intact Vascular, Medtronic, Mercator, Philips, Pluristem, Shockwave, Spectranetics, Terumo, TriReme, and Veryan.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Medtronic.