
Abstract
Aim:
Metabolic dysfunction-associated steatotic liver disease (MASLD) is a major cause of chronic liver disease, yet its pathogenesis remains incompletely understood. Oxidative stress is thought to play a key role in MASLD progression. This study aimed to investigate the role of cystathionine γ-lyase (CSE), an enzyme essential for cysteine and glutathione (GSH) biosynthesis, in MASLD development.
Results:
Choline-deficient high-fat diet (CDHFD) feeding led to elevated aspartate aminotransferase, alanine aminotransferase, hepatic triglyceride accumulation, vacuolization, macrophage infiltration, and cell death in both genotypes, with significantly greater changes observed in Cse−/− mice. CDHFD also reduced hepatic CSE expression in Cse+/+ mice and decreased cysteine/GSH levels in both genotypes, with more pronounced reductions in Cse−/− mice. Furthermore, Cse deletion was associated with increased oxidized glutathione/total GSH ratios and elevated levels of 4-hydroxynonenal and malondialdehyde. Expression of glutathione synthetase and γ-glutamyl transpeptidase was increased by CDHFD in Cse+/+ mice but blunted in Cse−/− mice. Furthermore, CSE deficiency exacerbated CDHFD-induced hepatic iron accumulation.
Innovation:
Our findings suggest that the CSE–cysteine–GSH axis may serve as a potential therapeutic target for MASLD, providing new intervention strategies beyond traditional approaches. This study provides new insights into the molecular mechanisms of MASLD and supports the development of antioxidant-based therapies.
Conclusions:
CSE deficiency exacerbates CDHFD-induced impairments of cysteine–GSH antioxidant axis, leading to hepatic oxidative stress and cell death. This indicates that CSE plays a protective role against MASLD development and progression. Antioxid. Redox Signal. 44, 11–23.
This is a visual representation of the abstract.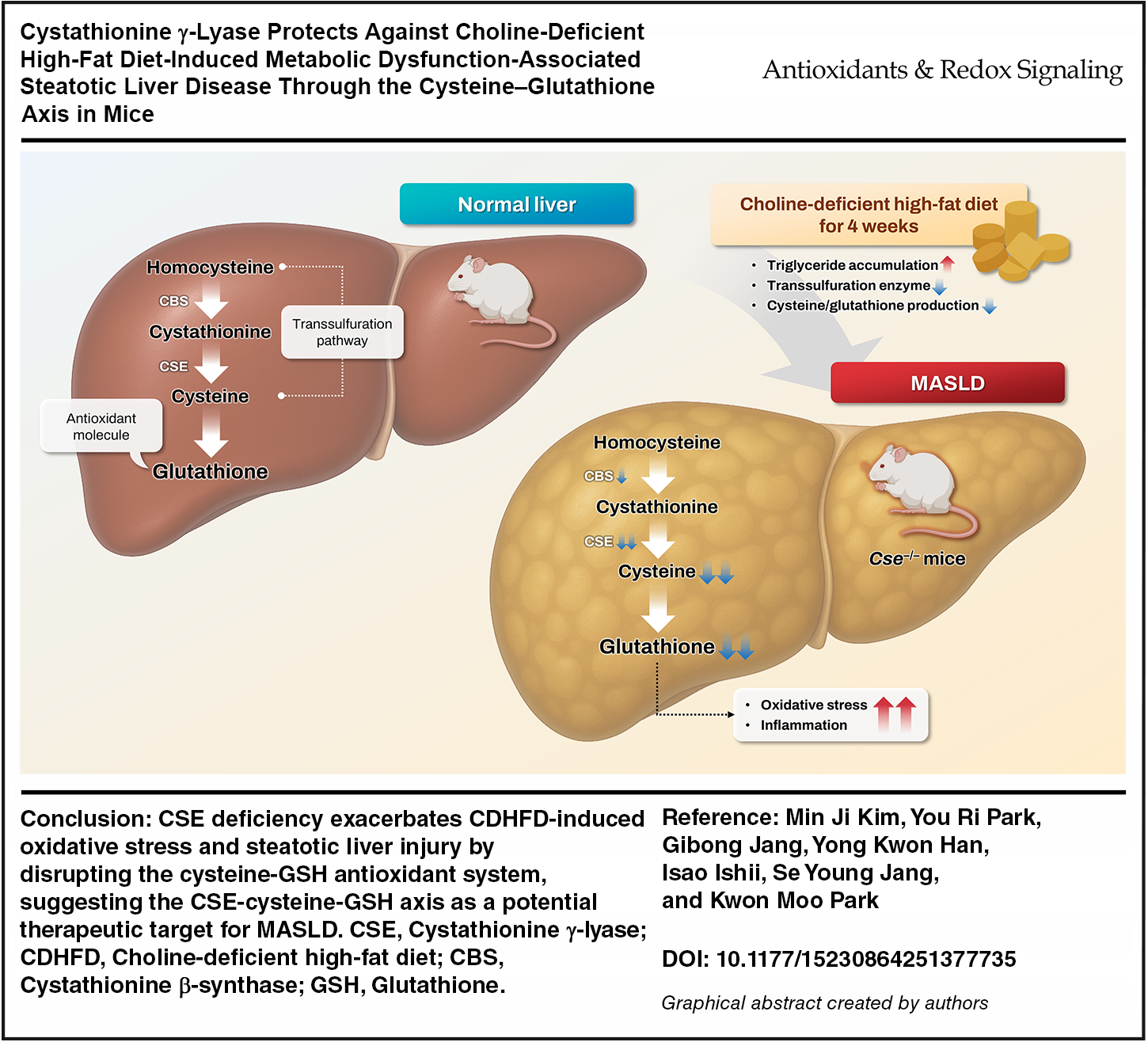
Keywords
Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD), is the leading cause of chronic liver disease worldwide (Younossi et al., 2023; Riazi et al., 2022). MASLD is characterized by excess fat accumulation in the liver, which can progress to metabolic dysfunction-associated steatohepatitis (MASH), a more severe subtype defined by steatosis, inflammation, and fibrosis (Rinella et al., 2023a). Furthermore, MASLD is closely associated with other metabolic disorders such as diabetes, hypertension, dyslipidemia, and cardiovascular disease (Targher et al., 2024; Chan et al., 2023). Although resmetirom has recently been approved as the first pharmacological treatment for nonalcoholic steatohepatitis (NASH) with fibrosis (Harrison et al., 2024), additional therapeutic options for MASLD are also needed. Thus, understanding the pathogenesis of MASLD is essential for developing targeted therapeutic and preventive strategies.
Innovation
Our findings suggest that the CSE–cysteine–GSH axis could be a potential therapeutic target for MASLD, offering a new avenue for intervention beyond traditional approaches that target inflammation or lipid metabolism. Furthermore, the identification of this pathway as a key player in MASLD pathology opens up innovative therapeutic possibilities. By targeting and enhancing this antioxidant system, new strategies for preventing and treating MASLD could be developed. This study contributes to a deeper understanding of the disease’s molecular mechanisms and provides a valuable foundation for future research aimed at developing antioxidant-based therapies for MASLD.
The liver plays a central role in lipid metabolism by regulating energy production and lipid transport to other organs (Rui, 2014). However, excessive fat accumulation disrupts normal fat metabolism, leading to exacerbation of mitochondrial fatty acid oxidation (Tilg and Moschen, 2010; Cusi, 2012; Zou et al., 2023). During this process, the electron transport chain interacts with oxygen, generating reactive oxygen species (ROS) (Zou et al., 2023; Chen et al., 2024). Excessive fatty acid oxidation results in increased ROS production within hepatocytes. When ROS production exceeds the antioxidant capacities of the hepatocytes, oxidative stress occurs (Tilg and Moschen, 2010; Cusi, 2012). This further impairs intrahepatic antioxidant defense systems, such as glutathione (GSH, the most abundant antioxidant molecule), leading to structural and functional cellular damage (Davies, 2000; Muriel and Gordillo, 2016). Numerous studies have suggested that oxidative stress is highly implicated in the development and progression of MASLD (Chen et al., 2020; Svobodova et al., 2025; Li et al., 2015).
GSH, which is composed of cysteine, glutamate, and glycine, is essential for maintaining redox homeostasis within cells (Vairetti et al., 2021; Yuan and Kaplowitz, 2009; Lu, 2009). GSH either scavenges intracellular ROS directly or supports the actions of other antioxidant molecules such as vitamins C and E, and antioxidant enzymes (Vairetti et al., 2021; Masella et al., 2005). The liver is the primary organ for maintaining body GSH homeostasis through its synthesis and degradation (Chen et al., 2024; Yuan and Kaplowitz, 2009). GSH acts as one of the most important antioxidants, protecting hepatocytes from oxidative stress (Vairetti et al., 2021; Yuan and Kaplowitz, 2009; Lu, 2009; Forman et al., 2009). Accumulating data have shown that defects in the GSH-associated antioxidant system contribute to the development of MASLD (Bin et al., 2017; Minetti et al., 2024; Svobodova et al., 2025).
The transsulfuration pathway is central to maintaining redox balance by linking methionine metabolism with cysteine and GSH biosynthesis. This pathway involves two key enzymes: cystathionine β-synthase (CBS), which catalyzes the condensation of homocysteine and serine to form cystathionine, and cystathionine γ-lyase (CGL, also known as CSE), which converts cystathionine into cysteine, α-ketobutyrate, and ammonia (Kabil and Banerjee, 2014). It has been known that CSE, a key transsulfuration enzyme in GSH homeostasis, is essential for cysteine production, which, together with glutamate and glycine, forms GSH (Werge et al., 2021; Diwakar and Ravindranath, 2007). Along with cysteine, CSE also produces hydrogen sulfide (H2S), a gasotransmitter with profound antioxidant effects (Werge et al., 2021). Several studies have reported that CSE protects the liver against NAFLD progression, primarily through H2S-mediated mechanisms (Ali et al., 2020; Xu et al., 2022).
Disruption of the transsulfuration pathway due to CSE deficiency may lead to homocysteine accumulation, a metabolite known to promote oxidative stress, endothelial dysfunction, and hepatocellular injury (Matte et al., 2009). Furthermore, recent evidence suggests that H2S functions as an epigenetic modulator of cytoprotective genes, and its deficiency may contribute to gene silencing, further exacerbating oxidative and metabolic stress (Dogaru and Munteanu, 2023).
However, the specific role of the CSE–cysteine–GSH axis in MASLD progression remains unclear. In this study, we investigate the involvement of the CSE–cysteine–GSH axis in MASLD progression by using Cse gene-deleted mice on a choline-deficient high-fat diet (CDHFD) model.
Results
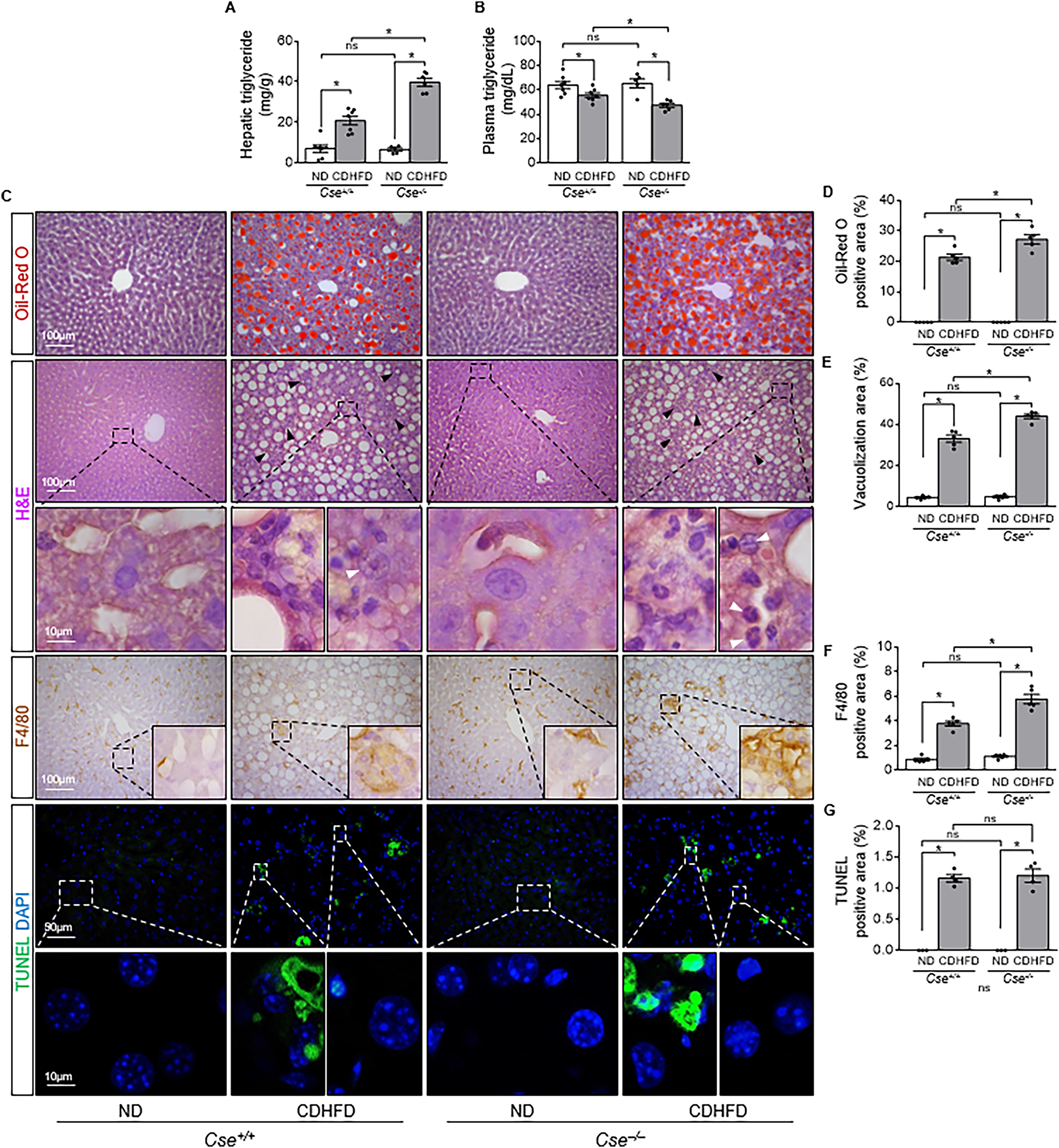
CSE gene deletion exacerbates CDHFD-induced hepatic lipid accumulation and macrophage infiltration
First, to decide the optimal CDHFD-feeding time, wild-type mice were fed either a normal diet (ND) or CDHFD for 2, 4, 6, and 8 weeks and then measured alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentration in plasma, and hepatic triglyceride (TG) levels. After CDHFD feeding, plasma ALT and hepatic TG levels started to increase at 2 weeks, peaked at 4 weeks, and then gradually declined (Supplementary Fig. S1A–C).
To investigate the role of CSE in MASLD development, Cse+/+ and Cse−/− mice were fed either an ND or CDHFD for 4 weeks. Daily caloric intake was higher in the CDHFD-Cse+/+ and CDHFD-Cse−/− mice compared with their respective ND groups (Supplementary Table S1). However, daily caloric intake was comparable between Cse+/+ and Cse−/− mice on either diet (Supplementary Table S1). Body weight was lower in both Cse+/+ and Cse−/− mice on the CDHFD compared with the ND, with no significant body differences between the genotypes (Supplementary Table S1). Plasma ALT and AST levels were elevated in both groups after CDHFD feeding, without significant differences between Cse+/+ and Cse−/− mice (Supplementary Table S1). In both Cse+/+ and Cse−/− mice, CDHFD increased TG concentrations in the liver, and decreased plasma TG levels (Fig. 1A, B). The hepatic TG increase in Cse−/− mice was greater than that in Cse+/+ mice, while the plasma TG decrease was more pronounced in Cse−/− mice (Fig. 1A, B). CDHFD also increased Oil-Red O-positive, vacuolization area, lobular inflammation (black arrows), and damaged hepatocytes (white arrows) in the livers of both Cse+/+ and Cse−/− mice compared with ND, with greater increases in the Cse−/− mice (Fig. 1C–E). Similarly, CDHFD expanded the F4/80-positive areas in both groups, with a larger expansion in Cse−/− mice (Fig. 1C and F). In addition, CDHFD induced hepatic cell death in both groups, with no significant differences between the Cse+/+ and Cse−/− groups (Fig. 1C and G). In wild-type mice, lobular inflammation (black arrows), hepatic vacuolization, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive signals were observed starting at 2 weeks after CDHFD and worsened over time (Supplementary Fig. S2A, B). Damaged hepatocytes (white arrows) were consistently observed at each time point (Supplementary Fig. S2A). These results indicate that CSE deficiency exacerbates CDHFD-induced MASLD.
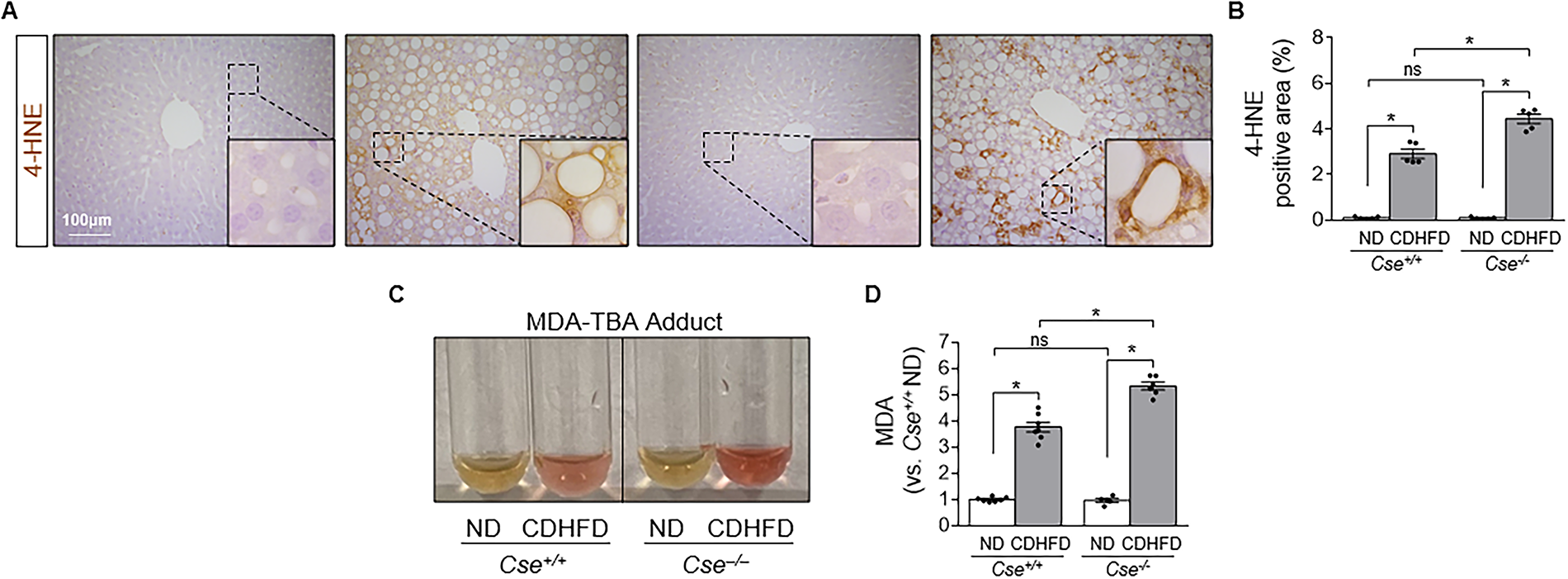
CSE deficiency exacerbates CDHFD-induced oxidative stress
As fat accumulation induces oxidative stress, which is a main contributor to liver damage, we measured lipid peroxidation products, including 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), in the liver of both Cse+/+ and Cse−/− mice after CDHFD feeding. CDHFD feeding significantly increased 4-HNE expression in both groups, with a greater increase in Cse−/− mice (Fig. 2A, B). 4-HNE was predominantly localized in the cytoplasm of hepatocytes in both Cse+/+ and Cse−/− mice after CDHFD feeding (Fig. 2A, B). MDA levels were elevated in the livers of both Cse+/+ and Cse−/− mice after CDHFD feeding, with a more pronounced increase in the Cse−/− mice than Cse+/+ mice (Fig. 2C, D). These results indicate that CDHFD induces oxidative stress in the liver, which is exacerbated by CSE deficiency.
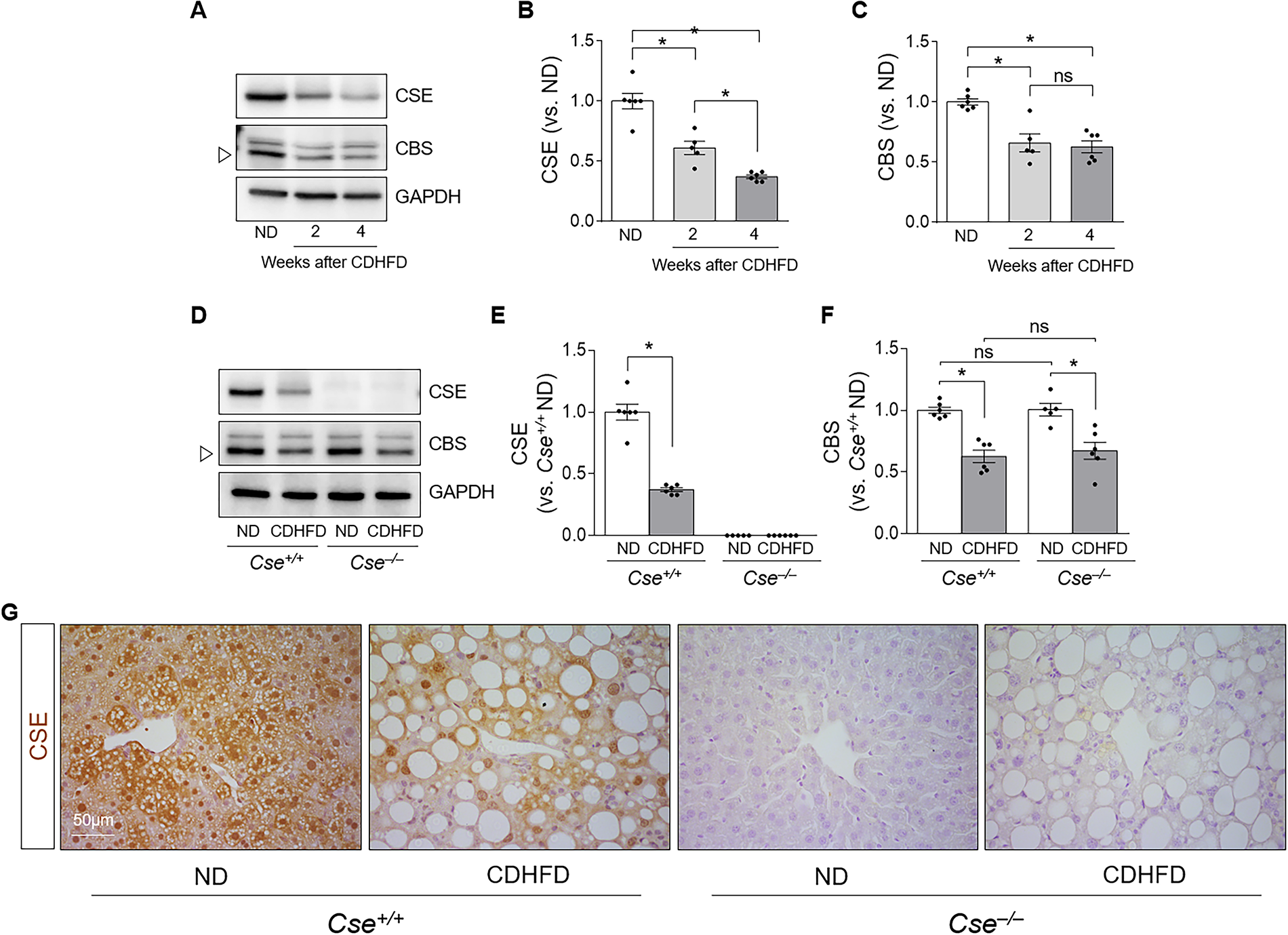
CSE deficiency reduces cysteine and GSH levels in the liver and aggravates CDHFD-induced reductions
CSE is essential for cysteine production, a component of GSH, the primary antioxidant molecule in the liver (Diwakar and Ravindranath, 2007). We investigated whether CDHFD feeding affects the expression of CSE and CBS, which produces cystathionine, a substrate of CSE. In wild-type mice, CSE and CBS expression decreased at 2 and 4 weeks after CDHFD feeding compared with ND (Fig. 3A–C). CSE expression was gradually decreased over time, but CBS expression showed no significant change at 2 and 4 weeks (Fig. 3A–C). CSE expression was absent in Cse−/− mice, and CDHFD reduced CSE expression in Cse+/+ mice compared with ND (Fig. 3D, E). CDHFD feeding also reduced CBS expression in both groups, with no significant differences between Cse+/+ and Cse−/− mice (Fig. 3D and F). CSE was predominantly observed in the cytoplasm and nuclei of liver cells in both ND and CDHFD feeding wild-type mice (Fig. 3G). These results indicate that CDHFD decreases both CSE and CBS expression in the liver.
We next determined the cysteine and GSH levels in the liver. Hepatic cysteine levels in ND-Cse−/− mice were approximately half those in ND-Cse+/+ mice (Fig. 4A). CDHFD decreased cysteine levels in both Cse+/+ and Cse−/− mice, with lower levels observed in Cse−/− mice (Fig. 4A). Both total GSH (tGSH, reduced GSH plus oxidized GSH) and oxidized GSH (GSSG) levels were significantly lower in the liver of ND-Cse−/− mice than in ND-Cse+/+ mice (Fig. 4B, C). CDHFD reduced tGSH levels in both groups (Fig. 4B), and increased GSSG levels in Cse−/− but not Cse+/+ mice (Fig. 4C). After CDHFD feeding, GSH levels in Cse−/− mice were about half of those in Cse+/+ mice (Fig. 4B). The GSSG:tGSH ratio was higher in ND-Cse−/− mice compared with Cse+/+ mice (Fig. 4D), and CDHFD increased the GSSG:tGSH ratio in both groups (Fig. 4D). The CDHFD-induced increase in GSSG:tGSH ratio (36.9% in Cse+/+ mice and 48.0% in Cse−/− mice, compared with the respective ND) was significantly greater than the increase in GSSG levels (4.7% in Cse+/+ mice and 14.3% in Cse−/− mice, compared with the respective ND). Consequently, the GSSG:tGSH ratio was found to be the highest in CDHFD-Cse−/− mice (Fig. 4D).
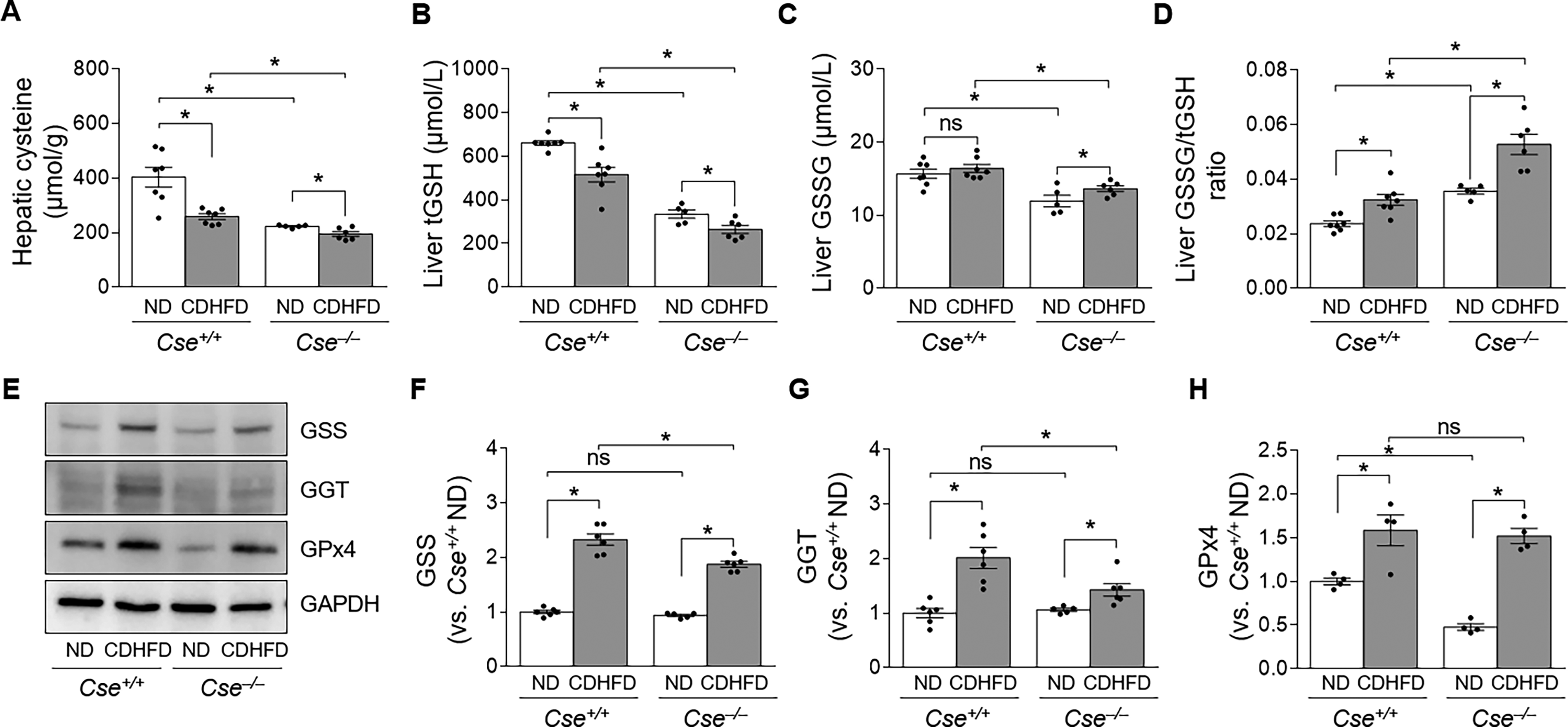
To further explore whether decreases in tGSH levels are associated with other GSH-related enzymes, we examined the expression of glutathione synthetase (GSS), γ-glutamyl transpeptidase (GGT), and glutathione peroxidase 4 (GPx4). There were no significant differences in GSS and GGT levels between ND-Cse+/+ and ND-Cse−/− mice (Fig. 4E–G). However, GPx4 levels were a significant difference between ND-Cse+/+ and ND-Cse−/− mice, in ND-Cse−/− mice they were lower than ND-Cse+/+ mice (Fig. 4E and H). CDHFD increased the expression of GSS in both groups, with a smaller increase observed in Cse−/− mice compared with Cse+/+ mice (Fig. 4E, F). Similarly, CDHFD feeding increased GGT expression in both Cse+/+ and Cse−/− mice, with a smaller increase in Cse−/− mice (Fig. 4E and G). Consistent with these findings, CDHFD feeding increased GPx4 expression in both Cse+/+ and Cse−/− mice (Fig. 4E and H). These results indicate that CDHFD feeding impairs the CSE–cysteine–GSH axis by increasing the GSSG:tGSH ratio and that Cse deletion further exacerbates this impairment.
CSE deficiency increases hepatic iron accumulation
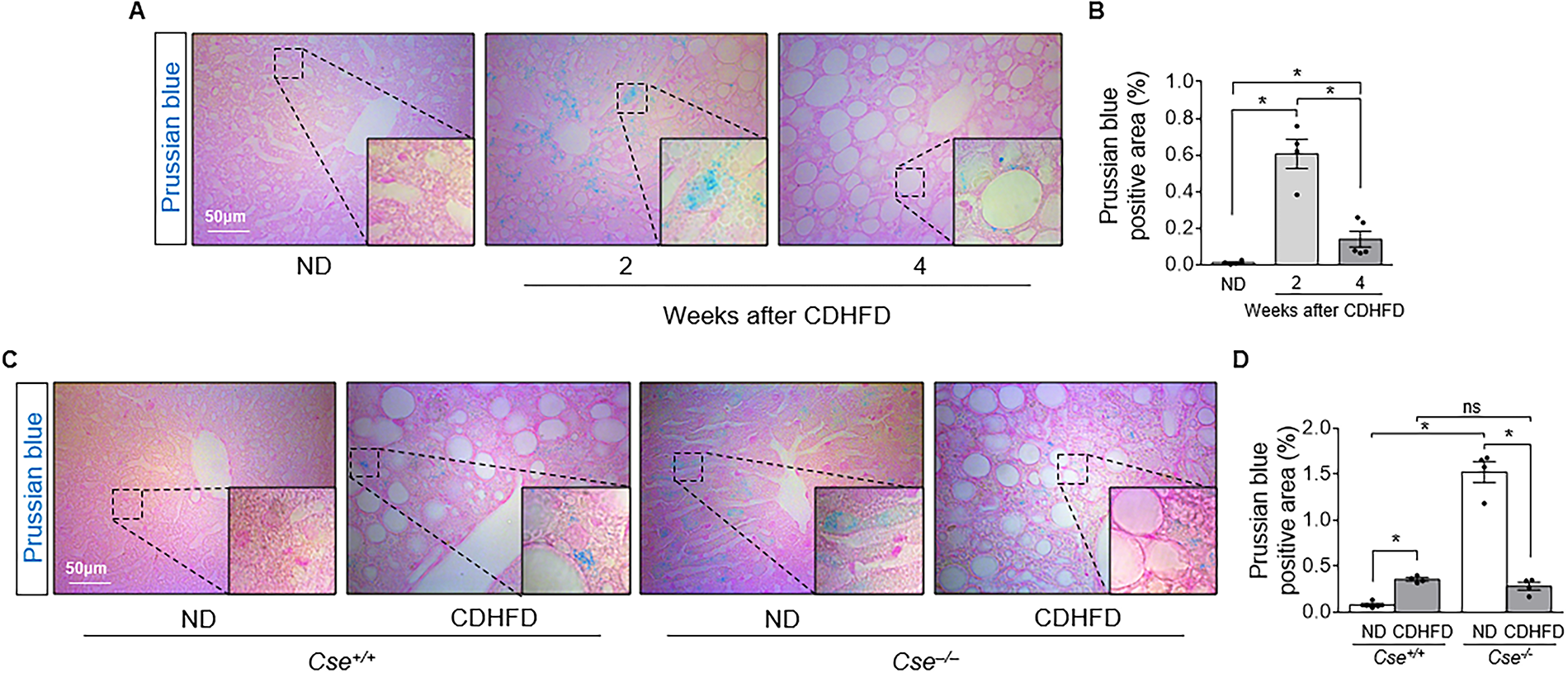
Since ferroptosis is an iron- and lipid peroxidation-dependent form of cell death (Li et al., 2020), we further investigated whether CDHFD-induced hepatocyte death is associated with iron accumulation in the liver by Prussian blue staining, which detects ferric iron (Fe3+). Prussian blue-positive signals were mainly observed in the cytoplasm of hepatocytes (Fig. 5A). In wild-type mice, Prussian blue-positive signals increased after 2 weeks of CDHFD feeding but decreased at 4 weeks, likely due to the reduced cytoplasmic area caused by lipid droplet expansion (Fig. 5A, B). Prussian blue-positive area was greater in ND-Cse−/− than in ND-Cse+/+ mice (Fig. 5C, D). After 4 weeks of CDHFD feeding, iron staining was substantially reduced in Cse−/− mice, whereas no reduction was observed in Cse+/+ mice (Fig. 5C, D). These findings indicate that CSE deficiency alters hepatic iron metabolism, leading to increased iron accumulation under ND conditions, which may contribute, at least in part, to the exacerbation of CDHFD-induced MASLD in Cse−/− mice.
Discussion
MASLD is a prevalent chronic liver disease, affecting over 30% of the global population (Rinella et al., 2023b; Yip et al., 2023). Despite extensive research into its pathogenesis, effective pharmacological therapies for MASLD remain limited (Chan et al., 2023; Tsochatzis and Noureddin, 2025). To better understand the molecular mechanisms of MASLD, various animal models have been developed, reflecting different etiologies (Lee et al., 2019). Among these, the high-fat diet (HFD) model is the most commonly used and well-characterized dietary animal model for MASLD studies (Fu et al., 2024). However, the HFD model has limitations, such as the diverse composition of diets and difficulties in inducing severe liver damage, including cell death, inflammation, and fibrosis (Fu et al., 2024; Yang et al., 2024).
To address the slow progression of MASLD in the HFD model, the methionine- and choline-deficient diet (MCD) was established. Although the MCD model induces MASLD more rapidly than the HFD model, it leads to severe weight loss, which does not accurately reflect the clinical condition of MASLD patients (Fu et al., 2024). In addition, methionine is a precursor of cysteine in the transsulfuration pathway (Bin et al., 2017; Distrutti et al., 2008), potentially confounding studies of the CSE–cysteine–GSH axis. To minimize the effects of methionine deficiency while enabling rapid MASLD induction, we used a CDHFD model.
Choline is essential for synthesizing very low-density lipoprotein (VLDL), which is crucial for lipid transport from cells to the bloodstream. Choline deficiency results in TG accumulation within hepatocytes due to impaired TG transport from the liver to the blood (Sugasawa et al., 2021; Yasuda et al., 2020). In addition, the excess fat provided by the HFD disrupts lipid metabolism, including fatty acid uptake and de novo fatty acid synthesis, resulting in increased free fatty acids and TG deposition in hepatocytes (Lian et al., 2020). Although choline deficiency affects extrahepatic systems—such as neurological function and homocysteine metabolism—hepatic steatosis remains the most prominent and reproducible phenotype, making choline-deficient diet models to be widely used in fatty liver disease research.
We acknowledge the limitations of the CDHFD model, including the reduced body weight of mice compared with the typical obese phenotype of MASLD patients. Nonetheless, we selected this model to induce MASLD more efficiently while avoiding the confounding effects of methionine deficiency associated with MCD models. To minimize systemic confounding, our analyses were confined to liver tissue. We focused on hepatic-specific oxidative stress markers—MDA and 4-HNE—to specifically evaluate liver oxidative injury associated with CSE deficiency in the context of steatotic liver disease.
Our findings demonstrate that CDHFD induces hepatic lipid accumulation, and CSE deficiency further aggravates this process, along with enhancing inflammation and oxidative stress compared with controls. CDHFD increased hepatic TG concentration (192.95% in Cse+/+ and 521.06% in Cse−/− mice compared with their respective ND groups, p < 0.05), while plasma TG concentrations decreased (12.95% in Cse+/+ and 26.64% in Cse−/− mice compared with their respective ND groups, p < 0.05). We therefore hypothesize that the increase in CDHFD-induced TG and lipid levels in the liver is caused by impaired TG release from the liver to the blood, combined with excessive lipid production within hepatocytes, with CSE playing a key role. Supporting this, Ali et al. reported that CSE inhibits hepatic lipid accumulation by promoting lipid secretion via ApoB (a major lipoprotein responsible for the assembly and transport of VLDL and LDL) and suppressing hepatic acetyl-CoA accumulation and fatty acid synthesis (Ali et al., 2020). Other studies have also shown that CSE deficiency aggravates HFD-induced lipid accumulation in hepatocytes (Loiselle et al., 2020; Sarna et al., 2015).
Excessive lipid accumulation induces ROS production and impairs cellular antioxidant defenses, resulting in oxidative stress (Cusi, 2012; Koek et al., 2011). Koruk et al. and Kumar et al. demonstrated that MDA levels are higher in patients with NAFLD than in healthy individuals (Koruk et al., 2004; Kumar et al., 2013). In our study, CDHFD increased lipid peroxidation, as indicated by elevated 4-HNE expression and MDA production in the liver, with CSE deficiency further exacerbating these effects. In addition, CDHFD increased F4/80-positive cells, particularly in regions with 4-HNE expression, and this increase was further exacerbated by CSE deficiency.
GSH, primarily synthesized from cysteine via the transsulfuration pathway, acts as a direct antioxidant to protect hepatocytes against ROS or pro-oxidants (Svobodova et al., 2025). Studies have shown that hepatic GSH levels are significantly lower in both NAFLD patients and animal models compared with healthy controls (Kumar et al., 2013; Dou et al., 2018; Grattagliano et al., 2008) and that GSH supplementation can alleviate NAFLD progression (Irie et al., 2016; Honda et al., 2017). CSE has been identified as a key enzyme in GSH synthesis (Badiei et al., 2019; Han et al., 2017), and we found that CDHFD reduced both GSH and cysteine levels, and Cse deletion augmented these CDHFD-induced reductions. Notably, Cse deletion alone reduced hepatic cysteine and GSH levels to approximately half of those in wild-type mice, and that CDHFD significantly decreased CSE expression more than CBS expression in the liver of Cse+/+ mice (62.84% and 37.36% of their respective ND levels), given that hepatic CSE expression is approximately 60-fold higher than CBS expression (Singh and Banerjee, 2011). These findings suggest a central role for CSE in MASLD progression. Consistently, reduced hepatic CSE and CBS expressions have also been reported in HFD-induced NAFLD animal models and human patients (Sarna et al., 2015; Sarna et al., 2016).
Several studies have shown that H2S, a gasotransmitter with pervasive signaling and antioxidant functions produced via the transsulfuration pathway (Sarna et al., 2015; Sun et al., 2021), is implicated in MASLD development. Xu et al. reported that hepatocyte‐specific CSE knockout decreased H2S generation by approximately 75%, while H2S supplementation attenuated HFD-induced NAFLD (Xu et al., 2022). Although we did not specifically assess the involvement of H2S in CDHFD-induced MASLD, these findings suggest that the protective effects of CSE are linked to both the cysteine–GSH antioxidant system and the antioxidant function of H2S.
Nevertheless, our focused analysis demonstrates that disruption of the cysteine–GSH axis plays a more direct and significant role in CDHFD-induced MASLD. CSE deficiency not only led to substantial reductions in hepatic cysteine and GSH levels, but also impaired the compensatory upregulation of GSS and GGT in response to oxidative stress. These changes were associated with elevated lipid peroxidation and hepatic inflammation, highlighting the CSE–cysteine–GSH axis as a central mediator of redox imbalance and MASLD progression.
Reduced GSH removes peroxides via GPx, converting GSH to GSSG, which is then reduced back to GSH by glutathione reductase (Lu, 2009). Therefore, the increased GSSG:tGSH ratio reflects increased oxidative stress and reduced GSH antioxidant capacity (Zitka et al., 2012). In our study, CDHFD increased GSSG levels by approximately 4.7% in Cse+/+ mice and 14.3% in Cse−/− mice, while the GSSG:tGSH ratio increased by 36.9% in Cse+/+ mice and 48.0% in Cse−/− mice. These results indicate that the increased GSSG:tGSH ratio is driven by reduced tGSH, further implicating the CSE–cysteine–GSH system in MASLD progression.
To compensate for the decreased intracellular GSH levels, cells activate alternative GSH-producing systems such as GSS and GGT to mitigate oxidative stress (Hayes and McLellan, 1999; Zhang et al., 2006). GSS is involved in the second step of GSH biosynthesis, regulating the condensation of γ-glutamyl-cysteine and glycine to produce GSH (Werge et al., 2021), while GGT plays a key role in the γ-glutamyl cycle, which governs GSH synthesis and degradation (Zhang and Forman, 2009). One study reported that fat accumulation in the liver induces hepatocellular damage and stimulates GGT synthesis (Hossain et al., 2016). GSS expression is also induced by pro-oxidants to increase antioxidant capacity (Lu, 2009; Lu, 2013).
In our study, although there were differences in tGSH levels between Cse+/+ and Cse−/− mice under ND conditions, GGT and GSS expression levels did not differ between the two groups. Moreover, CDHFD-induced increases in GGT and GSS expression were lower in Cse−/− mice. These results indicate that CSE deficiency alone does not affect GGT and GSS expression, but that tGSH reduction accompanied by increased oxidative stress can activate GGT and GSS expression to counteract oxidative stress. This is likely because cysteine is the rate-limiting precursor for GSH synthesis under oxidative stress, functioning both as the acceptor of the γ-glutamyl group in the GGT-catalyzed reaction (Hanigan, 2014) and as the substrate for GSS in GSH production (Lu, 2009; Bin et al., 2017). Therefore, we speculate that the reduced induction of GGT and GSS in Cse−/− mice following CDHFD feeding may be caused by lower cysteine levels. Consequently, lower GGT levels may limit GSH precursor delivery into cells, resulting in decreased GSH synthesis. While further studies are needed to define the role of cysteine in GSH production, our results suggest that CSE deficiency may impair compensatory increases in GGT and GSS under oxidative stress.
GPx4 is a major inhibitor of ferroptosis, and its reduction rapidly triggers this form of cell death (Li et al., 2020). Several studies have reported that elevated lipid peroxidation may induce GPx4 expression as a compensatory response (Qi et al., 2020). Similar to the results for GSS and GGT, CDHFD increased GPx4 expression. These findings suggest that lipid peroxidation can be both a cause and a consequence of GPx4 upregulation. Since GSH is essential for GPx4 activity (Ursini and Maiorino, 2020), the marked GSH depletion caused by CDHFD and further aggravated by CSE deficiency likely limits GPx4 function, thereby allowing lipid peroxidation to persist and contribute to ferroptosis.
In addition, we found that CDHFD increased hepatic iron accumulation, a well-recognized feature of NAFLD and a known promoter of ferroptosis, which may worsen liver damage and inflammation (Chen et al., 2022). Interestingly, CSE deficiency alone also resulted in increased hepatic iron accumulation. Consistent with the reduced GPx4 expression observed in ND-Cse−/− group, these results suggest that, although we did not determine how CSE deficiency affects hepatic iron levels under normal conditions, CSE deficiency enhances the susceptibility of liver to ferroptosis
In conclusion, our data demonstrate that the CDHFD impairs the CSE–cysteine–GSH system in the liver and that CSE deficiency exacerbates CDHFD-induced impairment of this antioxidant system, leading to aggravated oxidative stress and MASLD progression. These findings suggest that the CSE–cysteine–GSH axis could be a potential therapeutic target for MASLD. Future investigations should aim to directly compare the relative contributions of the CSE–cysteine–GSH and CSE–H2S pathways and evaluate therapeutic strategies targeting both. In addition, studies incorporating CSE activation, GSH supplementation, and iron modulation in MASLD models could provide further mechanistic and translational insights.
Materials and Methods
All experimental data were documented in bound paper laboratory notebooks. Data processing and visualization were performed using Microsoft Excel, GraphPad Prism, and PowerPoint. Electronic laboratory notebook was not used.
Animal experiments
All experiments were conducted using 9–10-week-old cse gene-deleted (Cse−/−) and wild-type (Cse+/+) male mice (22–25 g). The generation and characterization of Cse−/− mice have been described previously (Ishii et al., 2010). All procedures were approved by the Institutional Animal Care and Use Committee of Kyungpook National University, Republic of Korea (Approval No. KNU 2023–0285) and were performed in accordance with national regulations and institutional policies for animal welfare. Mice were randomly assigned to either an normal diet (ND 8.6 kcal% fat, SAFE®Diet Inc., Route de Saint-Bris, France) or a CDHFD group (60 kcal% fat, A06071302, Research Diets, New Brunswick, NJ, USA) for 4 weeks. Body weight and caloric intake were recorded weekly. At the end of the experiment, mice were euthanized with pentobarbital sodium (60 mg/kg, intraperitoneally), and livers and blood samples were collected for biochemical and histological analyses.
Blood chemistry
Blood was collected using a heparinized syringe. Plasma concentrations of ALT, AST, and TG were measured using a Vitro 250 Chemistry Analyzer (Johnson & Johnson, New Brunswick, NJ, USA).
Hepatic TG assay
TG concentrations in liver tissue were quantified using a commercial assay kit according to the manufacturer’s protocol (KTB2200, Abbkine Scientific Co, Ltd., Wuhan, China).
Histology
Paraffin-embedded liver tissues were sectioned at 3 μm thickness using a microtome (Leica, Bensheim, Germany). To remove the paraffin, the slides were placed in a 60°C oven for an hour, then immersed in xylene, and hydrated over 9 h through a graded ethanol series (100% to 80%). For H&E staining, the cytoplasm was stained with eosin for 1 min and washed with running water. Nuclei were stained with hematoxylin. Images were captured using a Leica microscope (DM2500, Leica, Wetzlar, Germany) and i-Solution software (IMT, Vancouver, Canada). Vacuolization areas (%) were analyzed using ImageJ software, with three fields per tissue from five mice.
Oil-Red O staining
Tissues embedded in the optimal cutting temperature compound were cut into 10-μm-thick sections and stained with Oil-Red O (Sigma-Aldrich, MO, USA) according to the manufacturer’s protocol. Sections were rinsed in distilled water for 5 min, treated with 60% isopropanol for 5 min, and then stained in Oil-Red O solution for 2 min. After rinsing in 60% isopropanol and distilled water for 5 min each, the sections were counterstained with hematoxylin. Images were captured using a Leica microscope and i-Solution software. Positive Oil-Red O-stained areas (%) were quantified using ImageJ software by analyzing three randomly selected fields per tissue sample from five mice.
Immunohistochemistry
Paraffin-fixed, 3-μm-thick sections were hydrated and permeabilized with 0.2% Triton-X for 5 min. Antigen retrieval was performed by immersing the slides in 10 mM sodium citrate buffer (pH 6.0) and autoclaving at 121°C for 10 min. Endogenous peroxidase activity was blocked by incubation in methanol containing 3% hydrogen peroxide for 30 min. The tissues were then immersed in PBS containing 5% bovine serum albumin (BSA) and incubated for 30 min at room temperature to remove nonspecific immunoreactivity in the tissue. The primary antibody was diluted in 1% BSA dissolved in PBS and reacted overnight at 4°C. After washing with PBS, the sections were incubated with the secondary antibody (diluted in 1% BSA dissolved in PBS) for an hour at room temperature. The slides were incubated with a 3′-diaminobenzidine substrate kit (Vector Laboratories, Burlingame, CA, USA) to develop color, before being counterstained with hematoxylin. The antibodies used for immunostaining were F4/80 (Cat no. MCA497GA, Bio-Rad, Hercules, CA, USA), 4-HNE (Cat no. ab46545, Abcam, Cambridge, MA, USA), and CSE (Cat no. sc-135203, Santa Cruz Biotechnology Inc., CA, USA). Positive staining areas (%) were quantified using ImageJ software by analyzing three randomly selected fields per tissue sample from five mice.
TUNEL assay
TUNEL staining in liver sections was performed according to the manufacturer’s protocol (11684795910, Roche, Basel, Switzerland).
Prussian blue staining
Liver sections were stained with Prussian blue staining according to the manufacturer’s protocol (AB150674, Abcam, Cambridge, MA, USA).
Western blotting analysis
To analyze protein expression, the frozen tissues were homogenized in a lysis buffer containing a protease inhibitor cocktail, a phosphatase inhibitor cocktail, and a HEPES buffer (hydroxyethyl piperazine ethane sulfonic acid buffer; pH 7.4). The homogenate was then centrifuged at 15,000 × g for 20 min at 4°C (Thermo Fisher Scientific, Vernon Hills, IL, USA). The supernatant was transferred to a sterilized tube and the protein concentration was measured using the bicinchoninic acid protein assay. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore, Burlington, MA, USA). The membranes were blocked with TBS-T (Tris-buffered saline with Tween-20) containing 1% BSA for an hour, and then incubated overnight at 4°C with primary antibodies diluted in TBS-T with 1% BSA.
After washing three times with TBS-T for 5 min each, membranes were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Blots were developed using an enhanced chemiluminescence system (Thermo Fisher Scientific), and protein bands were quantified using EvolutionCapt Edge software. The antibodies used in the Western blotting were CSE (Cat no. sc-135203, Santa Cruz Biotechnology Inc., CA, USA), CBS (Cat no. sc-133154, Santa Cruz), GSS (Cat no. 15712–1-AP, Proteintech, IL, USA), and γ-glutamyl transpeptidase (GGT; Cat no. sc-374495, Santa Cruz).
Measurement of tGSH and GSSG levels
Liver samples were homogenized in 5% sulfosalicylic acid (SSA; Sigma-Aldrich). The homogenates, consisting of 100 mg of liver tissue in 1 mL of 5% SSA, were centrifuged at 8000 × g for 10 min at 4°C. The supernatants were analyzed for tGSH and GSSG concentrations using the GSSG/GSH Quantification Kit (DojiNDo Laboratories, Kamimashiki-gun, Kumamoto, Japan) according to the manufacturer’s instructions.
Measurement of hepatic cysteine levels
Total cysteine concentrations in liver samples were measured using a Cysteine Assay Kit (MyBioSource, San Diego, CA, USA) according to the manufacturer’s instructions.
Measurement of MDA levels
MDA levels, indicative of lipid peroxidation, were measured by incubating samples with 2-thiobarbituric acid (Sigma-Aldrich), heating for 15 min, cooling, and centrifuging at 12,000 × g for 10 min. The absorbance of the supernatant was measured at 535 nm.
Statistical analysis
Data were analyzed using GraphPad Prism 9 software (GraphPad, San Diego, CA, USA). Data are presented as mean ± standard error of the mean. Comparisons between groups were made using Student’s t-test or one-way analysis of variance followed by Tukey’s post hoc test. Statistical significance was set at p < 0.05.
Authors’ Contributions
M.J.K.: Writing—original draft, methodology, conceptualization, investigation, and data analysis. Y.R.P.: Writing, methodology, conceptualization, and data analysis. G.J.: Methodology and conceptualization. Y.K.H.: Methodology, conceptualization, and data analysis. I.I.: Reviewing and editing the article. S.Y.J.: Writing the article, funding acquisition, conceptualization, data analysis, and supervision. K.M.P.: Writing the article, funding acquisition, conceptualization, data analysis, methodology, and supervision.
Footnotes
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work was supported by a Biomedical Research Institute grant, Kyungpook National University Hospital (2022). This research was also supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C0001).
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Statement
All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Kyungpook National University, Republic of Korea (Approval No. KNU 2023–0285), and carried out in compliance with national regulations and institutional policies for animal welfare.
Supplemental Material
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.