
Abstract
Although smoking-related interstitial lung diseases (SR-ILD) are a relatively rare group of entities, they are a relevant public health problem of growing importance, both because they affect young adults and because of their increasing prevalence in recent years due to increased tobacco consumption. In patients who smoke and have non-specific respiratory symptoms, SR-ILD should be ruled out, a term that encompasses a group of different entities in which the basis for diagnosis is the smoking history together with compatible respiratory functional findings, radiology and/or histology. An association has been established between tobacco smoke and a group of diseases that include respiratory bronchiolitis-associated interstitial lung disease (2%–3% of all ILD), desquamative interstitial pneumonia (<1%), Langerhans cell histiocytosis (3%–5%) and acute eosinophilic pneumonia. Smoking is considered a risk factor for idiopathic pulmonary fibrosis which has also been called combined fibroemphysema (5%–10% of all ILD); however, the role and impact of smoking in its development, remains to be determined. The likely interconnection between the mechanisms involved in inflammation and pulmonary fibrosis in all these processes often results in an overlapping of clinical, radiological, and histological features. In the absence of robust scientific evidence on its management, smoking cessation is the first measure to be taken into account. Although most diseases have a benign clinical course after smoking cessation, some cases may progress to chronic respiratory failure.
Introduction
Interstitial lung diseases (ILD) are a heterogeneous group of entities with distinctive clinical, radiological, functional and pathological features. Smoking is known to be a major public health problem and a major cause of morbidity and mortality. 1 While its association with entities such as lung cancer or chronic obstructive pulmonary disease is well documented, its relationship with other pathologies, such as smoking-related interstitial lung disease (SR-ILD), is less studied. 2

EVALI: e-cigarette or vaping use-associated lung injury; IIP: idiopathic interstitial pneumonias; ILA: interstitial lung abnormalities; ILD: interstitial lung disease; IPF: idiopathic pulmonary fibrosis; RA: rheumatoid arthritis; SRIF: smoking-associated interstitial fibrosis; VATS: video-assisted thoracic surgery biopsy. References:25–27,30.
Due to the diagnostic challenge of SR-ILD (Figure 1), we have carried out a review of the most frequent diseases in this group in daily clinical practice. Diagnostic algorithm for smoking-related interstitial lung diseases. Figure legend: AEP: acute eosinophilic pneumonia; AI: autoimmunity; BAL: bronchoalveolar lavage; CT: chest computed tomography; D: drugs; DIP: desquamative interstitial pneumonia; EVALI: e-cigarette or vaping use-associated lung injury; HP: hypersensitivity pneumonitis; ILA: interstitial lung abnormalities; ILD: interstitial lung disease; PH: pulmonary hypertension; PYI: pack-year index; LC: lung cancer; RB-ILD: respiratory bronchiolitis associated with interstitial lung disease; RF: risk factor; SRIF: smoking-related interstitial fibrosis.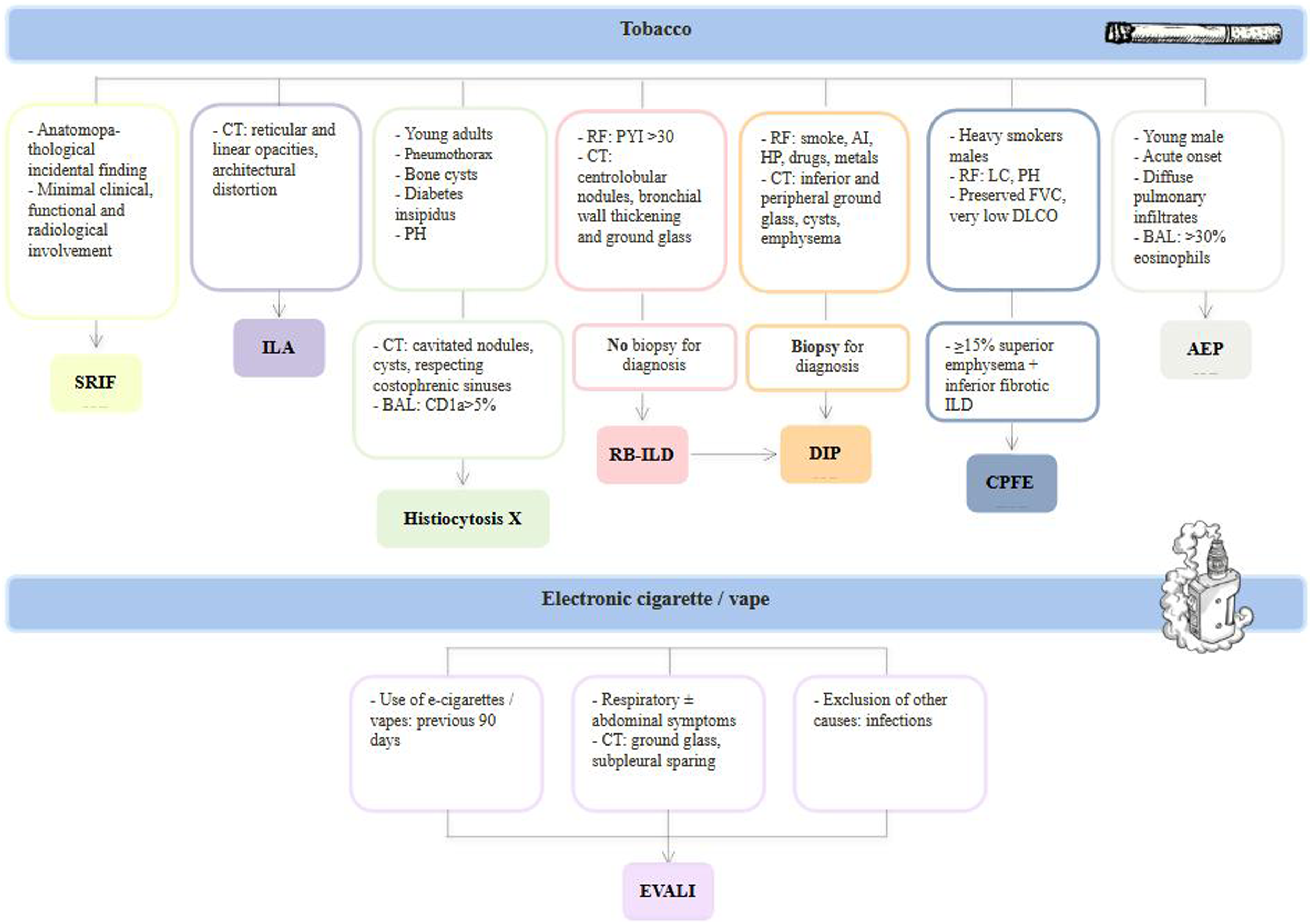
Methods
This is a narrative review article for the most relevant smoking-associated interstitial lung diseases. A selection has been made of the most important papers in routine clinical practice both for their frequency, particular specific characteristics and for a possible increased risk of progression due to their prognostic implications. For this purpose we conducted a literture search of Pubmed (Medline) and EMBASE using a predefined search strategy with a series of key words (“smoking”, “interstitial lung disease”); in addition, we also searched the “Cochrane Library” database. The search covered the period form June 1989 through December 2023 (period that was arbitrarily chosen). A total of 3000 articles were obtained, of which 100 were finally fully evaluated for this review.
Specific entities
Smoking-related interstitial fibrosis (SRIF)
Smoking-related interstitial fibrosis (SRIF) has been described by different authors under different names: respiratory bronchiolitis with interstitial pulmonary fibrosis or airspace enlargement with fibrosis.5,6 It is a recently described entity characterised by the presence of a fibrotic interstitial pattern with emphysema in smokers.
Histologically, SRIF has distinctive features that allow it to be differentiated from idiopathic pulmonary fibrosis (IPF) or other SR-ILD. Thus, at the tissue level, there is alveolar septal thickening with collagen deposition and minimal associated inflammation with subpleural and peribronchiolar distribution. Honey-combing is rare and pulmonary architecture is usually preserved. 7
Radiologically, various patterns can be seen including reticulation, micronodularity, ground glass and emphysema. 8 Thin-walled air cysts in mid and upper zones separated from the subpleural area have also been described, which differentiates it from usual interstitial pneumonia. 9 A key diagnostic feature is that the histological changes described are seen in several lung lobes, which does not always translate into radiological features in the same locations. This is because the interstitial fibrosis typical of SRIF does not cause architectural distortion and therefore does not necessarily cause radiological changes. The same is valid for emphysema (more frequently observed on histology than on radiology). 5
Most patients are usually clinically asymptomatic, as SRIF is usually an incidental histological finding. Respiratory function tests are preserved or show mild impairment in diffusion of carbon monoxide (DLCO). Although SRIF is usually an entity with a stable clinical course or minimal progression, 10 it can sometimes be associated with other ILDs with worse prognosis, such as IPF. 11
Pulmonary Langerhans cell histiocytosis (PLCH)
Pulmonary Langerhan’s cell histiocytosis (PLCH) is a rare disorder, almost exclusive to smokers or ex-smokers, characterised by clonal proliferation of Langerhans cells (antigen-presenting cells) and other inflammatory cells in the airways. 12 Initially, the pathogenesis was mainly attributed to a reactive process secondary to tobacco exposure. However, the paradigm shifted with the description of mutation BRAF proto-oncogene V600 E and mutations in genes resulting in activation of the MAPK (mitogen-activated protein kinase) pathway. 13 BRAF mechanism is of vital importance because of its potential therapeutic options, as is the one SR-ILD with evidenced based therapeutic interventions (see below in treatment section). Moreover, a combination of cigarette smoke and MAPK signaling pathway mutations results in a PLCH-like process in vitro. 14 Even though the precise mechanism of smoking in the pathogenesis remains unclear, it is likely that, in susceptible cases, tobacco acts as a pro-inflammatory trigger. 15
LCH is more common in young adults, has no gender predilection and can present with isolated pulmonary involvement (more common in adults) or with systemic manifestation. 16 In this line, the classification system differentiates between unifocal (solitary lesion) and single-system multifocal disease (>1 lesion). Multisystem LCH is characterized by involvement of ≥2 organs/organ systems.
Systemic LCH presents (SLCH) in a continuum of organic involvement, ranging from a solitary eosinophilic granuloma to widespread disseminated disease with organ dysfunction. The skeleton is the most commonly affected system, as bone lesions are present in approximately 80% of patients with SLCH, and in half of them, lesions are single. Other organs that may be affected include: skin (especially in childrens), hematopoietic (cytopenias indicate a poor prognosis), spleen, liver, central nervous system, lymph node and others (i.e. thyroid, thymus, etc). Adult SLCH could present in association with neoplastic diseases, especially other myeloproliferative neoplasms.
On the other hand, the clinical presentation of Pulmonary Langerhans cell histiocytosis is non-specific (exertional dyspnoea and cough) and half of the patients have normal lung function. Cases with functional involvement may show both an obstructive and restrictive pattern with decreased DLCO. 17 Several helpful diagnostic features are associated with PLCH like pneumothorax (15%–20% of cases and in relation with cystic lung disease), bone cysts and diabetes insipidus. Pulmonary hypertension is a serious and frequent complication (40% of all patients) that manifests in advanced cases and its severity is not linearly correlated with interstitial involvement, but rather with vascular involvement, which must be assessed in each case. 18
Radiologically, in the early stages of PLCH there are usually cavitated nodules predominantly in the upper fields that later coalesce and give rise, in more advanced stages to cysts. Typical cysts of this entity are homogeneous in size and respect the costo-phrenic sinuses (which differentiates them from those seen in lymphangioeliomyomatosis).
Characteristic for bronchoalveolar lavage (BAL), is the presence of CD1a + cells >5% which states high specificity as oppose to sensitivity. 19 In cases requiring biopsy for definitive diagnosis, bronchiolocentric cellular nodules, bronchiolar wall destruction, inflammation, cysts, emphysema and even fibrosis will be observed. Birbeck granules are characteristic (elongated, zipperlike cytoplasmic structures measuring 200-400 × 33 nm viewed under electron microscopy) and immunohistochemistry with CD1a and CD207+ is specific for the diagnosis. 12
The most important therapeutic measure is smoking cessation, which usually stabilises the course of the disease. Other treatments with little scientific evidence and poor response have been used, such as corticosteroids, cladribine (cytotoxic), vasodilators (if pulmonary hypertension) or vemurafenib (for BRAF - V600 E mutation). 20 This latter treatment differentiates PLCH from other SR-ILD by being able to have a specific target. In severe cases, lung transplantation is an option, although the disease may recur. 18 The natural history of the disease is variable and ranges from a favorable evolution (50%), stability (30%–40%) and the development of recurrent pneumothoraces (10%–20%) that may progress to chronic respiratory failure. 21
Respiratory bronchiolitis - associated ILD (RB-ILD)
Respiratory bronchiolitis - associated ILD (RB-ILD) is an ILD that corresponds to the anatomopathological lesion of respiratory bronchiolitis due to smoking. Respiratory bronchiolitis presents with asymptomatic accumulation of pigmented macrophages in the respiratory bronchioles of smokers, whereas RB-ILD is symptomatic and has radiological and functional manifestations typical of SR-ILD. It is slightly more frequent in males with a cumulative tobacco consumption of more than 30 packs/year. Common radiological findings are ill-defined centrolobular nodules, ground glass opacities, emphysema in upper lung fields and bronchial wall thickening. 22 Respiratory function impairment may be restrictive, mild obstructive and/or have DLCO impairment. BAL is non-specific and may show smoker’s characteristic macrophages. Biopsy is not usually required for diagnosis and pathological lesions also reveal aggregates of brown pigmented macrophages together with patchy peribronchiolar infiltrates. 23
Some authors postulate that RB-ILD is part of the initial clinical spectrum of desquamative interstitial pneumonia (DIP), while others question this, as DIP is not exclusive to smokers. 23
The mainstay of treatment for RB-ILD secondary to smoking is cessation, reserving the use of corticosteroids for cases where the disease progresses despite smoking cessation. If no response is obtained, immunosuppressive therapy has been used in some cases. 23 The prognosis is generally favourable after smoking cessation and fibrotic progression has not been reported. 24
Desquamative interstitial pneumonia (DIP)
Main differences between respiratory bronchiolitis associated with interstitial lung disease (RB-ILD) and desquamative interstitial pneumonia (DIP).
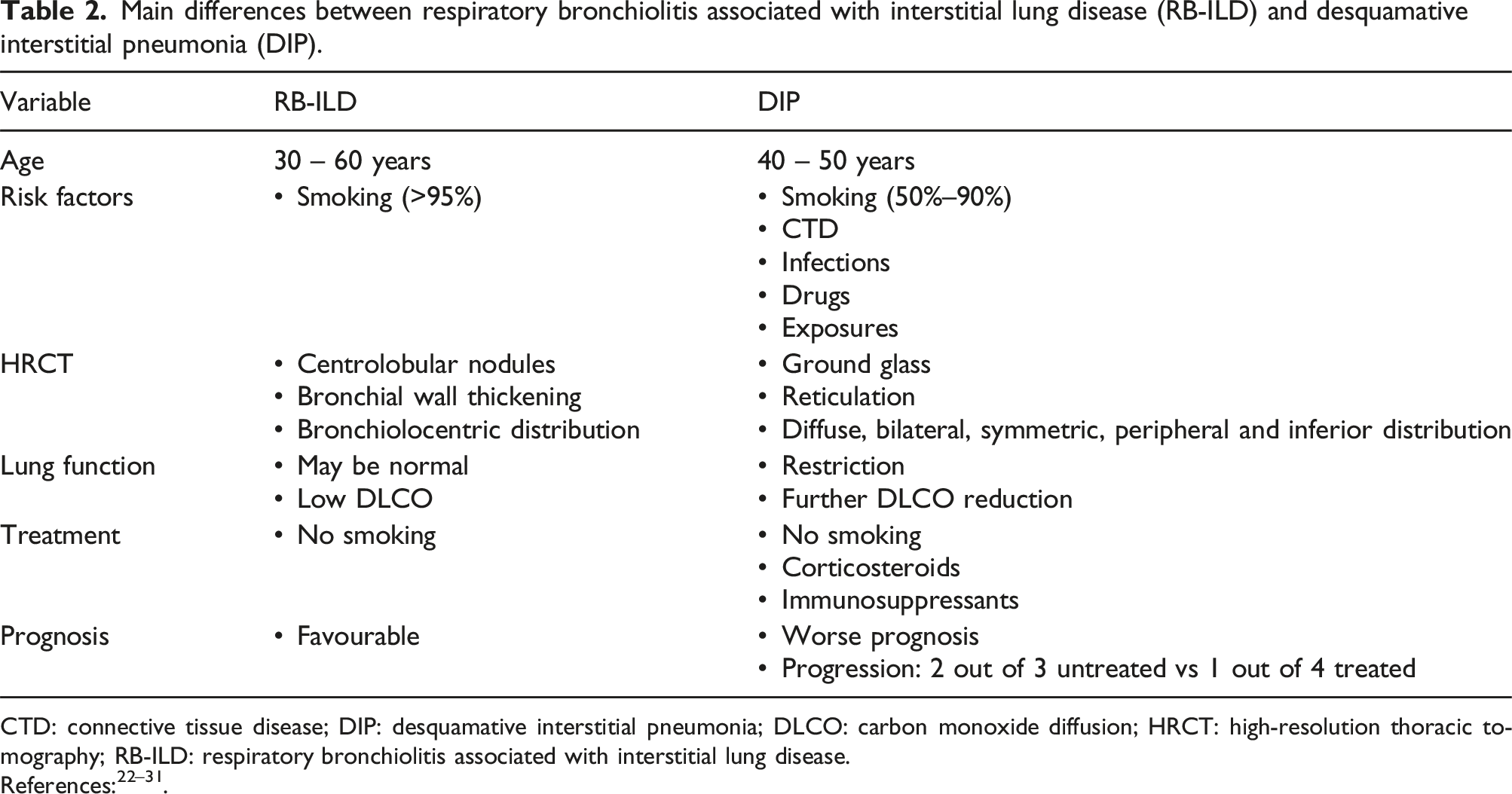
CTD: connective tissue disease; DIP: desquamative interstitial pneumonia; DLCO: carbon monoxide diffusion; HRCT: high-resolution thoracic tomography; RB-ILD: respiratory bronchiolitis associated with interstitial lung disease.
DIP is more common in males in the 4th-6th decade of life 26 and the reduction in DLCO is more marked than the decrease in lung volumes on pulmonary function tests. In thoracic HRCT, bilateral, symmetric, peripheral, inferior and patchy or diffuse ground-glass, opacities together with a greater or lesser degree of emphysema and/or cystic lesions is most prevalent. In more advanced cases, traction bronchiectasis or even honey-combing may be observed. 27
Anatomopathologically, lung architecture is usually preserved and there is a discrete chronic interstitial inflammatory infiltrate together with the observation of smoker’s characteristic macrophages in the alveolar spaces. 28 The main distinguishing feature between RB-ILD and DIP is that the latter has uniform and diffuse involvement and is not bronchiolocentric like the former. BAL in DIP is usually macrophagic, but may show eosinophilia in a minority of cases. 29
The mainstay of DIP treatment is smoking cessation. There is no robust evidence on the use of other treatments such as corticosteroids, which may stabilise the disease but do not improve it, or immunosuppressants (azathioprine or cyclophosphamide). 30 In two out of three untreated patients and one out of four treated patients, the disease will progress. 30 However, fibrotic progression is rare and 10-years survival is around 70%. 31
Fibroemphysema
In combined pulmonary fibrosis and emphysema syndrome (CPFE), patients have a combination of at least 15% emphysema with inferior pulmonary fibrosis. 32 It is most prevalent in men aged 60-80 years, heavy smokers and with associated comorbidity [lung cancer (especially epidermoid) and pulmonary hypertension in up to 47%–90% of cases], which causes higher morbidity and mortality. 32 As mentioned, smoking is the main risk factor, but there are others, such as agricultural exposures. One of the characteristic features of the entity is the relative preservation of lung volumes (due to the presence of emphysema which counteracts the loss of respiratory function of fibrosis) together with a marked impairment of DLCO (decreased by both emphysema and interstitial fibrotic involvement). 33
As the name suggests, thoracic HRCT will show emphysema and bullae predominantly in the upper lung fields, as well as fibrotic changes (honey-combing, reticulation, traction bronchiectasis and/or architectural distortion) in the lower lung fields. The most common radiological patter is usual intestitial pneumonia (UIP), but others, such as nonspecific interstitial pneumonia (NSIP) or DIP, may occur. A clinical course with frequent exacerbations may represent a poor prognosis with a gradual progression to chronic respiratory failure.
The most important therapeutic measure is abstinence from smoking together with optimisation of emphysema and fibrosis treatment. Bronchodilators may also be useful if there is airflow limitation. Anti-fibrotic drugs should be considered in cases with progressive fibrosis. 34 To date, there are no clinical trials, or strong scientific evidence for its management.
E-cigarette or vaping use-associated lung injury (EVALI)
The term E-cigarette or vaping use-associated lung injury, better known by the acronym EVALI, has been combined to describe the damage induced by e-cigarettes or vapers. As its name suggests, it is the consequence of interstitial damage produced by tobacco derivatives (tetrahydrocannabinoids and various nicotine derivatives). 35 Since 2019, 2500 cases of EVALI requiring hospital admission had been published in the United States, with 55 associated deaths. 36 EVALI is more common in young men (mean age of presentation 45 years) and its aetiopathogenesis is not known exactly, although it has been postulated that vitamin E acetate, an ingredient in e-cigarettes, is related to its development, as high titres of this vitamin have been found in BAL. 37
The clinical presentation is non-specific (dyspnoea, cough, chest pain, constitutional symptoms or fever) and its main feature is the presence of abdominal symptoms (in up to 80% of cases). Bilateral ground-glass opacities are seen on thoracic HRCT, which involve the subpleural area. Diagnosis requires a history of e-cigarette use in the previous 90 days, characteristic radiology and ruling out other possibilities (mainly infectious origin). 38 There are as yet no clinical trials or other robust scientific evidence for its management. There are case series describing treatment with corticosteroids and antibiotics with good results if there is respiratory failure. 39
Interstitial lung abnormalities (ILA)
Due to the widespread use of thoracic HRCT, more and more radiological alterations are being described in smokers who do not meet diagnostic criteria of any defined entity, such as interstitial lung abnormalities (ILA). As a consequence of the recent implementation of lung cancer screening, different interstitial lung abnormalities have started to be observed in smokers, even in the absence of respiratory symptoms. 40 Although its clinical significance is unclear, ILA has been associated with respiratory functional impairment, and some authors postulate that it may be an early stage of IPF, as radiological and/or respiratory functional progression is observed in these patients. 41 Incipient fibrotic changes such as reticulations, linear opacities or architectural distortion (parenchymal irregularities, vascular tortuosity and/or anatomical alterations) are frequently observed on HRCT. These radiological alterations have been correlated with histological involvement in smokers’ biopsies. It should be noted that the ATS 2022 IPF guideline, with respect to its previous version, removes the concept of “early-UIP” from the radiological pattern “indeterminate for UIP” in order not to confuse it with the term ILA. 34
ILA is an entity with no specific treatment except smoking cessation. The importance of follow-up has been described in case fibrotic progression develops and in case re-classification to other interstitial entities such as IPF is required. 42 ILA is an independent prognostic factor for mortality, with up to 40% of patients showing radiological progression after 4-6 years. 34
Acute eosinophilic pneumonia (AEP)
Acute eosinophilic pneumonia (AEP) is an acute-onset respiratory disease (dyspnoea, cough, fever and chest pain of a few days or weeks’ duration) characterised by diffuse bilateral pulmonary infiltrates, respiratory failure and an elevated BAL eosinophil count. 43 It predominates in young male patients and its main risk factor is smoking. Other less consistent associations are the use of e-cigarettes, drugs, pharmaceuticals or occupational exposures. 44
As mentioned, the eosinophil count is key to the diagnosis. At least in its initial presentation, there is usually no peripheral eosinophilia, although it may appear later in the course of the disease. In contrast, in BAL there is a high number of eosinophils (>25%) which, in the right context, is usually sufficient for diagnosis. Interestingly, although not fully explained, the presence of a higher number of eosinophils in BAL is associated with a lower degree of respiratory failure, 45 when the opposite would be expected.
Chest radiograph shows bilateral alveolar and interstitial opacities together with Kerley B lines. Bilateral pleural effusion may be present in half of the patients. Thoracic HRCT shows ground-glass opacities, consolidations and interlobular septal thickening. 46 Histologically, infiltration of the alveoli, bronchioles and interstitial space by eosinophils is observed, with oedema and diffuse alveolar damage. 47
Treatment of AEP is based on avoiding smoking and administration of corticosteroids with a gradual tapering of the dose over 2-4 weeks. With an early approach, the prognosis is usually satisfactory and most patients experience complete resolution. Relapses are rare, which differentiates it from its chronic form (chronic eosinophilic pneumonia). A thorough assessment of the different sources of exposure is recommended in order to achieve appropriate avoidance behaviour. 48
Other SR-ILD where smoking is a risk for its pathogenesis and frequency
Idiopathic pulmonary fibrosis (IPF)
IPF has a smoking prevalence of over 70% (there is a high prevalence of smoking but the disease pathogenesis is complex). It is more common in men over 50 years of age. The characteristic radiological pattern is UIP, although a probable UIP pattern may be diagnostic if other causes are excluded. BAL may show mild neutrophilia and eosinophilia, and histology will show involvement of the lower lung zones and subpleural fields with patchy fibrosis, architectural distortion, fibroblastic foci and heterogeneous involvement (alternation of affected areas with normal lung). Nintedanib anti-fibrotic treatment is successful in halting the decline in lung function and transplantation is reserved for more advanced cases. 34 In this line in INBUILD trial, patients with fibrotic ILD (other than IPF) who received nintedanib had a slower rate of progression than those who received placebo, independent of the fibrotic pattern on high-resolution CT. 49 At the moment more studies are needed to be able to make a recommendation for Pirfenidone in fibrotic ILD non IPF.
Rheumatoid arthritis associated interstitial lung disease (RA-ILD)
Rheumatoid arthritis-associated ILD (RA-ILD) is a complex entity, both because of its frequency and potential severity, and because of the difficulty involved in its therapeutic management. Its incidence increases in smokers. The published evidence is scarce, with low methodological quality and contradictory results. In more than three quarters of patients, the onset of ILD occurs after the diagnosis of RA; UIP histological pattern is the most frequent and represents the second most common cause of death in patients with RA. A particularly difficult aspect is its treatment as these patients are systematically excluded from most clinical trials and, in addition, some of the drugs used for RA may worsen or trigger ILD (as Methotrexate). 50
Alveolar proteinosis
Up to 56%–79% of cases of alveolar proteinosis have a history of current or past smoking habit. In severe cases, the treatment of choice is total lung lavage and as a second line, if anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) antibodies are demonstrated, inhalation therapy with GM-CSF can be evaluated. 51
Goodpasture’s syndrome
A consistent association between smoking and the development of Goodpasture’s syndrome with alveolar haemorrhage has been described. Tobacco smoke is therefore considered the main risk factor for this entity. Treatment in the acute phase is complex and requires corticosteroids, immunosuppressants, plasmapheresis or even haemodialysis in cases with severe renal failure. 52
Conclusions
SR-ILD are a heterogeneous set of individual entities with common features due to their clinical, radiological and/or histologic overlap, which poses a great challenge in the differential diagnosis between diseases.
The key to diagnosis is based on three fundamental pillars: a history of smoking, thoracic HRCT images and histology (in cases where this is necessary). It is also important to grade the radiological and functional involvement, which will determine the prognosis. It is known that the interpretation of hitological findings is complex, since it is often not known whether they are incidental tissue alterations, without clinical correlation, or whether they really represent incipient data that will condition a future progression towards an SR-ILD. For all these reasons, they constitute a group of diseases in which a multidisciplinary approach is particularly relevant for their correct diagnosis and management.
New studies are needed to answer the large number of questions they raise, as only by better understanding their characteristics, evolution and possible treatments will we be able to improve the prognosis of patients with SR-ILD.
Footnotes
Author contributions
A.C., Author and drafting. Conception and design. Approval of the final manuscript. J.S.A., Co-author. Approval of the final manuscript. V.R., Co-author. Approval of the final manuscript. L.F., Co-author. Approval of the final manuscript. N.R-N., Co-author. Approval of the final manuscript. M.E.T., Co-author. Approval of the final manuscript. L.V., Author and drafting. Conception and design. Approval of the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.