
Abstract
Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a rare disease, idiopathic in most of the cases described in the literature. We report the case of a 55-year-old patient, non-smoker with tuberculosis treated in his youth, starting with progressive dyspnoea and cough, with radiographic abnormalities consisting of pleural thickening, bronchiectasis and structural distortion predominantly in the upper lobes. Due to functional impairment and persistent radiographic abnormalities, biopsy by video-assisted thoracoscopic surgical was decided. The presence of striking elastosis, absence of interstitial damage and abrupt boundary of the fibrous lesion with healthy lung allowed the diagnosis of IPPFE. Currently, the patient has no specific treatment and is in follow-up in the Transplant Unit.
Keywords
Introduction
Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a rare disease, recently described as an entity; the first description appeared in Japanese literature in 2004, 1 followed by the inclusion of this disorder in the American Thoracic Society/European Respiratory Society classification of idiopathic interstitial pneumonias (IIPs) in 2013 as a rare IIP. 2
Diagnosis of the disease is based on clinical presentation. There are respiratory symptoms of interstitial pneumonic processes: recurrent airway infections, chronic cough and dyspnoea on exertion, with or without weight loss, but there may be a silent period during which patients have no symptoms. There are radiologic images (bilateral apical pleural thickening, volume loss of upper lobes due to reticular and nodular abnormalities including thickening of interlobular septa and at later stages honeycombing) and histopathological findings (severe fibrosis of the visceral pleura, prominent subpleural fibroelastosis composed of dense collagen and elastic fibres and abrupt transition of fibroelastosis to the healthy lung parenchyma).
The differential diagnosis includes other causes of pleural thickening with fibrosis such as radiation-induced lung injury, scarring, pneumonic processes or interstitial pneumonia in fibrotic phase.
So far, diagnosis of disease occurs in advanced stages when pleural fibroelastosis is already developed, therefore long-term studies are needed to understand the evolution of this disease over time.
Case report
A 55-year-old male, non-smoker without any occupational exposure was referred to pulmonology department for the management of suspected interstitial lung disease observed on chest radiography and chest CT in 2011, after being diagnosed with autoimmune negative anterior uveitis. He complained of progressive dyspnoea (grade I–II of Medical Research Council (MRC)) and non-productive cough. He had a history of bronchiectasis secondary to pulmonary tuberculosis 30 years ago, as well as oesophageal spasm as his only other relevant history. His physical examination revealed bilateral basal crackles but otherwise unremarkable.
Lung function test in his first consultation were as follows: forced expiratory volume in 1 second (FEV1), 60% predicted; forced vital capacity (FVC), 58% predicted; FEV1/FVC, 84%; diffusing capacity of the lungs for carbon monoxide (DLco), 62% predicted; DLco/alveolar volume (VA), 110% predicted; total lung capacity (TLC), 74% predicted; and residual volume, 120% predicted. Blood gas parameters were within the reference range values: pH 7.41; partial pressure of arterial oxygen, 85 mmHg; partial pressure of arterial carbon dioxide, 40 mmHg; oxygen saturation, 98%.
Chest radiography (Figure 1) and chest CT (Figure 2) showed bilateral apical pleural thickening associated with bronchiectasis, parenchymal bands, distortion and shrinkage in both upper lobes. The left lower lobe was also affected in a similar way.
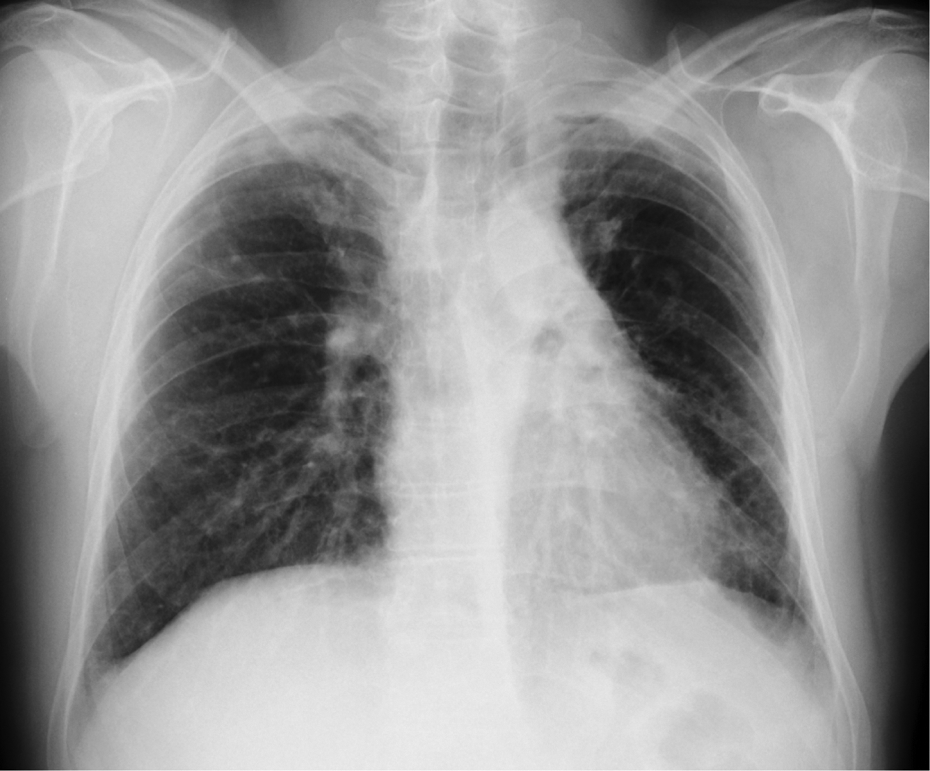
Image of chest radiography where bilateral upper lobe volume loss, thickening and parenchymal bands can be observed as well as a left basal reticulation pattern with mediastinal pleural affectation.

Images of axial HRCT. (a) This image shows diffuse pleural thickening with bilateral focal subpleural consolidations, architectural distortion and traction bronchiectasis accompanied by volume loss in the upper lobes. (b) HRTC image at the right middle lobar bronchus level which demonstrates the same findings in left lower lobe and less involvement on the right side with minimal pleural thickening and middle lobe bronchiectasis. (c) Image of coronal multiplanar reconstruction where dense apical thickening with bronchiectasis and upper lobe volume loss can be perceived.
Given the non-diagnostic radiological pattern and progressive decline in his respiratory function tests, it was decided to perform a video-assisted thoracoscopic surgical lung biopsy of the right upper lobe. The diagnosis of IPPFE was confirmed histopathologically by the appearance of severe subpleural fibrosis associated with septal elastosis (Figure 3), after ruling out Mycobacterium tuberculosis infection or fungus with microbiological study.

Picture package containing histologic features and a surgical lung biopsy which evidence marked pleural thickening, subpleural fibrosis with abrupt transition of fibroelastosis and unaffected lung parenchyma. (a) Image of lung tissue stained with Masson’s trichrome which shows significant elastosis (×2). (b) Elastic van Gieson staining that shows a large amount of short, curled, amorphous and randomly oriented elastic fibres (×10). (c) Image of elastic van Gieson staining (×40).
In 2013, the patient was referred to a Lung Transplant Centre, where he is currently under periodic reviews. A progressive and significant clinical and functional impairment (−20% in FVC in 4 years), with development of exertional hypoxaemia have been observed.
His last lung function test shows: FEV1, 41% predicted; FVC, 38% predicted; DLco, 39% predicted; DL/VA, 103% predicted; with significant reduction in TLC (55% predicted). He walked 450 meters in the six-minute walk test (6-WT) with final oxygen desaturation to 78%. For some months, now he requires oxygen to walk, (grade III dyspnoea of MRC, with poor effort tolerance).
For all these reasons, he has been included on the waiting list for lung transplantation.
Discussion
IPPFE is a rare disorder described in 2004, characterized by presenting pleural thickening, loss of volume of upper lobes and prominent subpleural fibroelastosis. The histopathological features are similar to those of idiopathic pulmonary upper lobe fibrosis (IPUF), as previously described in Japanese literature. 3 Currently, IPUF and IPPFE are considered within the same entity. Recently, IPPFE was included in the classification of the group of idiopathic interstitial pneumonias. 2
Both the pathological and aetiological mechanisms are as yet unknown, with a 10%–30% of cases being idiopathic. 4 Regarding secondary mechanisms, late complication of bone marrow/haematopoietic stem cell transplantation or a manifestation of chronic lung rejection in lung transplant recipients have been described. 5 It has also been described in relation with collagen vascular disease, chemotherapy (10% of cases) and even as a familial case. 6 Finally, the possible relation with recurrent respiratory infections (particularly Aspergillus species) is also being studied due to the chronic inflammation that occurs in these entities. 1 It seems that there is a female predominance with a mean age of 50 at the time of presentation. The onset of the disease is not related to smoking.
There is a variety in clinical presentation, the most common being, as in our patient, dyspnoea and dry cough (91% and 50% of patients, respectively) but also the occurrence of spontaneous pneumothorax or pneumomediastinum 7 as the most striking form of presentation.
Following the onset of symptoms in this case, chest CT was performed, which revealed bilateral apical pleural thickening and subpleural fibrosis associated with signs of upper lobe fibrosis, sparing the lower lobes. Bronchiectasis could initially be interpreted in the context of tuberculosis suffered by the patient in the past, as in other cases, 8,9 but the appearance of fibrosis raised the possibility of interstitial fibrosis associated with connective tissue pathologies (due to uveitis, which was eventually discarded) or an atypical form of usual interstitial pneumonia. 10 In addition, the differential diagnosis also included other pleurofibrotic entities such as chronic hypersensitivity pneumonitis, advanced sarcoidosis or Langerhans cell histiocytosis 11 and even the so-called apical cap, 12 a localized lesion of subpleural fibrosis, which usually occurs in the upper lobes and tends to affect mature smokers; it is typically asymptomatic and does not progress.
Given the progressive deterioration of respiratory function and non-diagnostic pattern seen on the radiological imaging, it was decided to perform a pleuropulmonary biopsy, which gave the final diagnosis as is the approach in most published cases where the final diagnosis was also made on biopsy. 13 However, the risk of developing pneumothorax (bullae develop along the course of the disease), 14 which can get complicated with bronchopleural fistula, 15 makes some authors suggest that for future cases the diagnosis of IPPFE should be done without lung biopsy based solely on the radiological characteristics, symptoms and some biomarkers that may be useful for this. 16 In our patient, the biopsy was diagnostic and helped to rule out infectious causes such as Aspergillus infection or active tuberculosis.
There is no specific treatment for this disease; corticosteroids, immunosuppressants, and even N-acetylcysteine have been tested 17 with mixed results, that is, either ineffective, with transient effect or even with zero effect. There are also cases, such as ours, in which no treatment was given. 18 Mean survival of the disease is 11 years; it is thought to be more aggressive in familial cases 6 and even in idiopathic cases. In cases of progression, lung transplantation is proposed as a last option, 14 although to date there has been only three cases reported, two of which had prior exposure to chemoteraphy. 6,19
Further studies are warranted to see both the natural course of the disease and the therapeutic options that may be effective.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.