
Abstract
Although glucagon has been shown to exert pleiotropic actions in various types of cells and organs through the interaction with its receptor, its pathophysiological role in atherosclerotic cardiovascular disease remains unclear. Here, we examined whether and how glucagon could attenuate the progression of atherosclerotic plaques in apolipoprotein E-deficient mice (ApoE−/−), an animal model of atherosclerosis. Glucagon (138 or 413 nmol/kg/day) or vehicle was infused to mice at 16 weeks of age. After 4-week treatment, vascular samples were collected for histological and RT-PCR analyses. Human monocytic THP-1 cells were pre-incubated with or without a glucagon receptor antagonist L-168049, and then treated with or without glucagon for 7 h. Gene and protein expressions were determined by RT-PCR and western blot analyses, respectively. High-dose glucagon infusion significantly decreased aortic plaque area and volume in ApoE−/− mice, both of which were inversely correlated with plasma glucagon levels. Glucagon infusion also reduced the ratio of pro-inflammatory interleukin-1β to anti-inflammatory interleukin-10 gene expression in aortae. Glucagon receptor was expressed in THP-1 cells, and 1 nM glucagon decreased the ratio of interleukin-1β to interleukin-10 gene expression, which was significantly prevented by L-168049. Our present findings suggest that glucagon could exert atheroprotection partly via its anti-inflammatory property.
Introduction
Glucagon is a peptide hormone secreted from pancreatic alpha cells in response to plasma levels of glucose and amino acids.1,2 Classically, glucagon and insulin are thought to exert opposing effects on glucose and lipid metabolisms1,2; glucagon not only stimulates gluconeogenesis, glycogenolysis, and ketogenesis in the liver, but also promotes lipolysis in the adipose tissues.1,2 Therefore, glucagon has long been used for the treatment of hypoglycemia in diabetic patients.1,2 In addition, there is accumulating evidence to show that glucagon has pleiotropic actions in various types of cells and organs through the interaction with glucagon receptor.1–3
In addition to the above-mentioned physiological stimuli for glucagon section, plasma level of glucagon could be affected by oral hypoglycemic agents.1,2 Indeed, sodium-glucose transporter 2 (SGLT2) inhibitors has been reported to increase plasma glucagon level,4,5 which may attenuate the glucose-lowering action of SGLT2 inhibitors. Furthermore, given the ketogenic effect of glucagon,1,2 it could also be involved in the increased risk of normoglycemic ketoacidosis in type 2 diabetic patients treated with SGLT2 inhibitors. 6 However, the effects of glucagon beyond metabolic and anthropometric parameters remain largely unknown.
Treatment with SGLT2 inhibitors is recommended for type 2 diabetic patients with a high risk for or a history of atherosclerotic cardiovascular disease. 7 Since the risk of atherosclerotic cardiovascular disease is increased in patients with type 2 diabetes,8,9 it is a critical issue to clarify the pathophysiological role of glucagon in atherosclerotic cardiovascular disease. In this study, we examined whether and how glucagon could attenuate the progression of atherosclerotic plaque in apolipoprotein E-deficient (ApoE−/−) mice, an animal model of atherosclerosis. 10
Materials and methods
Agents
Chemical agents were obtained as follows: human glucagon and liraglutide from Novo Nordisk Japan (Chiyoda, Tokyo, Japan); ethylenediaminetetraacetic acid (EDTA) and aprotinin from Fuji Film WAKO (Osaka, Osaka, Japan); L168049 from Merck Japan (Meguro, Tokyo, Japan); exendin (9-39) amide from ANASPEC (Fremont, CA, USA).
Animal experiments
Design of animal experiments was approved by the Animal Care Committee of Showa University School of Medicine (approval numbers: 09058), and all experiments were conducted under strict adherence to the Guide for the Care and Use of Laboratory Animals as previously described. 10 Male ApoE−/− mice (BALB/c. KOR/StmSlc-Apoeshl) at 6 weeks of age were purchased from Sankyo Labo Service (Edogawa, Tokyo, Japan), and maintained on standard rodent chow with free access to water. Mice at 12 weeks of age were switched to high-cholesterol western diet feeding (F2HFD1: fat, 36% of total kcal; cholesterol, 1.25% w/w; Oriental Koubo, Itabashi, Tokyo, Japan). After 4 weeks of pre-feeding with high-cholesterol western diet, mice at 16 weeks of age were randomly assigned to the following three treatment groups: vehicle, low-dose glucagon (138 nmol/kg/day), or high-dose glucagon (413 nmol/kg/day). 11 Acid buffer (pH 3.5) was used as a solvent to increase solubility of glucagon. 1 Glucagon was continuously infused to mice with osmotic pumps implanted in the dorsal subcutaneous tissue (Alzet minipump 1002; Cupertino, CA, USA). 10 The pumps were replaced every 14 days to avoid glucagon degradation. After 4 weeks of treatment with vehicle or glucagon, mice were euthanized by overdose of isoflurane, and blood and vascular samples were collected for biochemical, histopathological, and reverse-transcription polymerase chain reaction (RT-PCR) analyses. Heart and liver were weighted to obtain heat and liver indexes, which were calculated by dividing tissue weight by body weight.
Measurement of plasma levels and blood pressure
Blood samples were collected after 6 h of fasting, and immediately mixed with EDTA and aprotinin to avoid glucagon degradation (final concentrations of each reagent were 1 mg/ml and 500 U/ml, respectively). Plasma glucose and lipid levels were determined by enzymatic electrode and colorimetric assay as previously described. 10 Non-high-density lipoprotein (Non-HDL) cholesterol level was calculated by subtracting HDL cholesterol level from total cholesterol level. Plasma levels of glucagon, insulin, IL-1β, and IL-10 were measured by enzyme-linked immunosorbent assay (ELISA) kits (Glucagon ELISA Kit, Mercodia Inc, Winston Salem, NC, USA; Ultra-sensitive insulin ELISA, Morinaga Milk Industry Co. Ltd., Yokohama, Japan; Mouse IL-1β Duoset ELISA and Mouse IL-10 Duoset ELISA, R&D Systems, Minneapolis, MN, USA). A kit for glucagon can detect both mouse and human glucagon. Systolic blood pressure and pulse rates were measured using the non-invasive tail-cuff method as previously described. 10
Assessment of atherosclerotic plaques
Atherosclerotic plaques on the aortae were evaluated as previously described. 10 Aortae from the aortic sinus to the bifurcation of common iliac arteries were longitudinally dissected and stained with oil red O to evaluate en face plaque formation (plaque area). Cyrosections of the aortic sinus were stained with oil red O and picro-sirius red (PSR) and immunostained with anti-MOMA-2 antibody (Abcam Japan, Chuo, Tokyo, Japan; Product ID: ab33451; RRID:AB_776518; dilution: 1:60) to assess lipid deposition (plaque volume), intraplaque collagen content, and intraplaque macrophage accumulation levels, respectively. 10 The images were analyzed using Image J software (National Institutes of Health, Bethesda, MD, USA).
Cell culture experiments
Human monocytic THP-1 cells (cell number: JCRB0112; authentication: HLA-DR, My 7, My 9, OKM1 positive; nonspecific esterase positive; peroxidase negative; a-naftol ASD chloroacetate esterase negative) were obtained from JCRB Cell Bank of National Institutes of Biomedical Innovation, Health and Nutrition (Ibaraki, Osaka, Japan), and cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS). Cells were seeded onto 24-well plates at the cell density of 1.5 × 105 and 5.0 × 105 per well for RT-PCR and ELISA, respectively. After serum starvation overnight, cells were pre-treated with or without L-168049, an antagonist of glucagon receptor at 300 nM or exendin (9-39) amide, an antagonist for glucagon like peptide-1 (GLP-1) receptor at 100 nM for 1 h,12,13 and then stimulated with or without glucagon at 1 nM or liraglutide at 100 nM for 7 h for RT-PCR analysis. Serum-starved cells were stimulated with or without glucagon at 10 nM for 24 h for ELISA.
Western blot analysis
Protein expression levels were determined by western blot analysis as previously described. 10 In brief, 5 μg of extracted proteins was electrophoresed in polyacrylamide gels and transferred to polyvinylidene fluoride membranes. The membranes were incubated with blocking reagent for 1 h, primary antibodies overnight, and then secondary antibody for 1 h. The antibodies were used with the following concentrations: rabbit polyclonal antibody raised against glucagon receptor (Alomone labs, Jerusalem, Israel; Product ID: AGR-024, RRID: AB_2340975; dilution: 1:10,000), monoclonal antibody raised against β-actin (Santa Cruz Biotechnology, Dallas, TX, USA; Product ID: sc-47778; RRID: AB_2714189; dilution: 1:1000), and donkey polyclonal antibody raised against anti-rabbit IgG conjugated with horseradish peroxidase (GE Healthcare Japan; Product ID: NA9341; RRID: AB_772206; dilution: 1:40,000).
Real-time RT-PCR
Gene expression levels were determined by RT-PCR as previously described. 10 In brief, total RNA was extracted from tissues and cells to synthesize cDNA. Quantitative real-time RT-PCR was performed using the TaqMan gene expression assay (Life Technologies Japan, Minato, Tokyo). The following pre-designed TaqMan probe sets were used for the assay: interleukin-1beta (Il-1β), Mm00434228_m1; mouse Il-10, Mm00439614_m1; human IL-1β, Hs01555410_m1; human IL-10, Hs00961622_m1. Gene expression levels of target molecules were normalized with those of the housekeeping gene 18S ribosomal RNA (18srna, Mm03928990_g1; 18sRNA, Hs 99999901_S1), and the data were presented as relative levels to the controls.
ELISA
Cell culture medium where THP-1 cells were grown were collected after 24 h-treatment with vehicle or glucagon. Then the IL-10 levels were measured with ELISA kit according to the manufacturer’s instruction (Human IL-10 DuoSet ELISA, R&D Systems).
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Statistic comparisons were conducted using JMP software (version 13; SAS Institute Inc., NC, USA). Unpaired t-test was used for the comparisons of two groups; One-way ANOVA followed by Tukey’s test was used for the comparisons of three or more groups; correlation was tested by Pearson correlation coefficient. The significance level was defined as p < 0.05.
Results
Effects of chronic glucagon infusion on atherosclerotic plaques in ApoE−/− mice
Vehicle, low-dose glucagon, or high-dose glucagon was continuously infused to ApoE−/− mice for 4 weeks. As shown in Figure 1(a), plasma glucagon levels were dose-dependently increased by a continuous infusion of glucagon. Metabolic parameters are shown in Table 1. There were no significant differences in biochemical and anthropometric parameters except for plasma glucose levels among the groups; although plasma levels of glucose among the three groups were within a normal range, they were modestly lower in high-dose glucagon group than vehicle group. Plasma levels of insulin, total cholesterol, high-density lipoprotein (HDL)-cholesterol, non-HDL-cholesterol, triglycerides, and free fatty acids were comparable among the groups. As shown in Table 1, plasma IL-10 levels were significantly higher in high-dose glucagon group than those of vehicle group. However, plasma IL-1β levels in all groups were below the detectable limit of ELISA kit used here (<16 pg/ml).
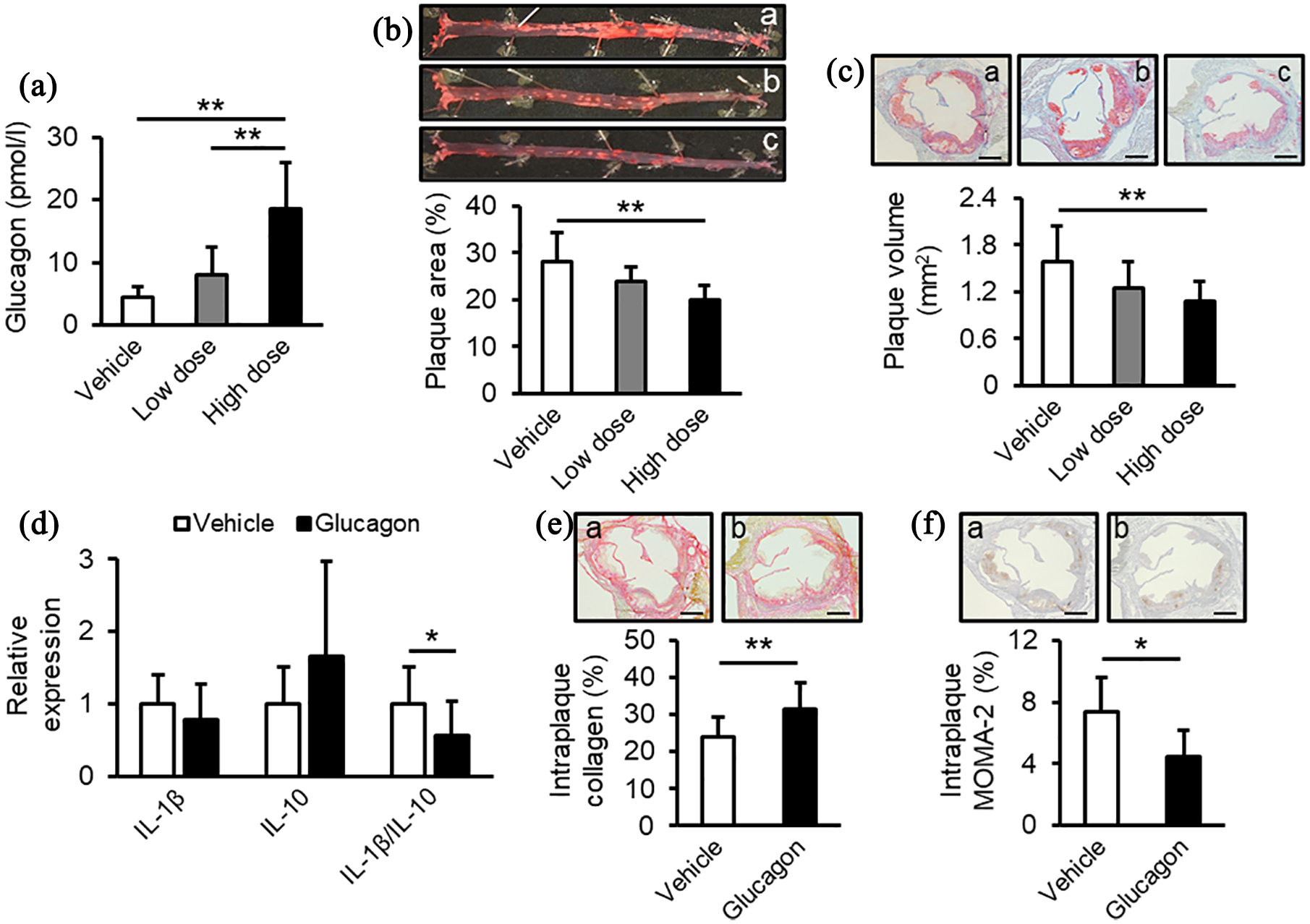
Anti-atherogenic effects of chronic glucagon infusion in ApoE−/− mice. ApoE−/− mice fed with high-cholesterol diet were treated with vehicle, low-dose glucagon (138 nmol/kg/day), or high-dose glucagon (413 nmol/kg/day) for 4 weeks. (a) Plasma glucagon levels in the end of the experiment. (b) Plaque area on the aortic surface. Upper images show representative images of the aorta with oil red O. a, Vehicle; b, low-dose glucagon; c, high-dose glucagon. (c) Plaque volumes at the aortic sinus. Upper images show representative images of the aortic sinus with oil red O. a, Vehicle; b, low-dose glucagon; c, high-dose glucagon. (d) Gene expression levels of Il-1β and Il-10 and expression ratio of Il-1β to Il-10. Gene expression levels of target molecules were normalized with those of the housekeeping gene 18S ribosomal RNA, and the data were shown as relative levels to the control mice. (e and f) Intraplaque collagen content (e) and macrophage accumulation levels (f) at the aortic sinus. Upper images show representative images of the aortic sinus with PSR. a, Vehicle; b, glucagon. Vehicle, n = 11; low-dose glucagon, n = 10; high-dose glucagon; n = 10. d–f: Mice treated with low-dose and high-dose glucagon were combined as the glucagon group.
Clinical characteristics of ApoE−/− mice treated with vehicle, low-dose glucagon, or high-dose glucagon.
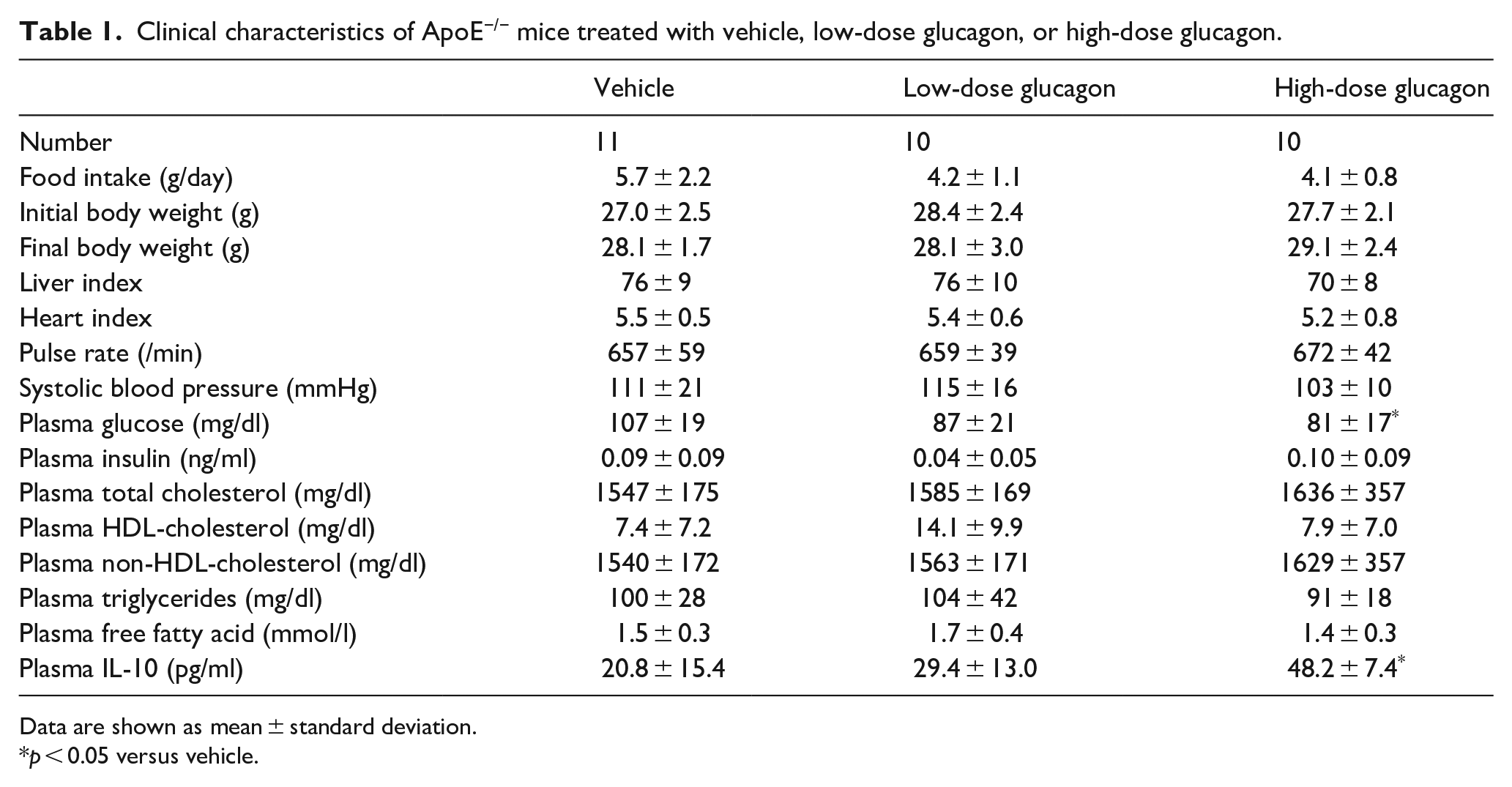
Data are shown as mean ± standard deviation.
p < 0.05 versus vehicle.
As shown in Figure 1(b) and (c), atherosclerotic plaque areas on the aortic surfaces and plaque volumes at the aortic sinuses were significantly reduced by a high-dose of glucagon infusion, both of which were inversely correlated with plasma levels of glucagon after 4-week intervention (Pearson’s correlation coefficient: p = 0.02, r = −0.44 for both). In addition, although gene expression levels of pro-inflammatory cytokine Il-1β or anti-inflammatory cytokine Il-10 in the right brachiocephalic arteries, an atherosclerosis-prone site, 14 were not affected by the treatment with glucagon, a continuous infusion of glucagon significantly decreased the gene expression ratio of Il-1β to Il-10 compared with vehicle (Figure 1(d)). Furthermore, glucagon treatment significantly increased collagen content and reduced macrophage accumulation within the atherosclerotic plaques compared with vehicle as well (Figure 1(e) and (f)).
Effects of glucagon and liraglutide on Il-1β and Il-10 gene expression in cultured human THP-1 cells
Western blot analysis revealed that human monocytic THP-1 cells expressed glucagon receptor, whose expression levels were approximately 60% of those of the liver, a target organ of glucagon (Figure 2(a)). Glucagon at 1 nM significantly increased gene expression levels of IL-10 without affecting those of IL-1β, thereby resulting in the decreased ratio of IL-1β to IL-10, all of which were prevented by the treatment with L-168049, an antagonist of glucagon receptor at 300 nM (Figure 2(b)). L-168049 at 300 nM alone did not affect gene expression levels of IL-1β or IL-10 in THP-1 cells. Consistent with gene expression levels of IL-10, glucagon at 10 nM significantly stimulated the production of IL-10 released from THP-1 cells (Figure 2(c)). Liraglutide, an agonist of GLP-1 receptor mimicked the effects of glucagon in THP-1 cells (Figure 2(d)), whereas exendin (9-39) amide, an antagonist of GLP-1 receptor did not affect the effects of glucagon on gene expression levels of IL-1β or IL-10 in THP-1 cells, irrespective of the presence or absence of glucagon (Figure 2(e)).
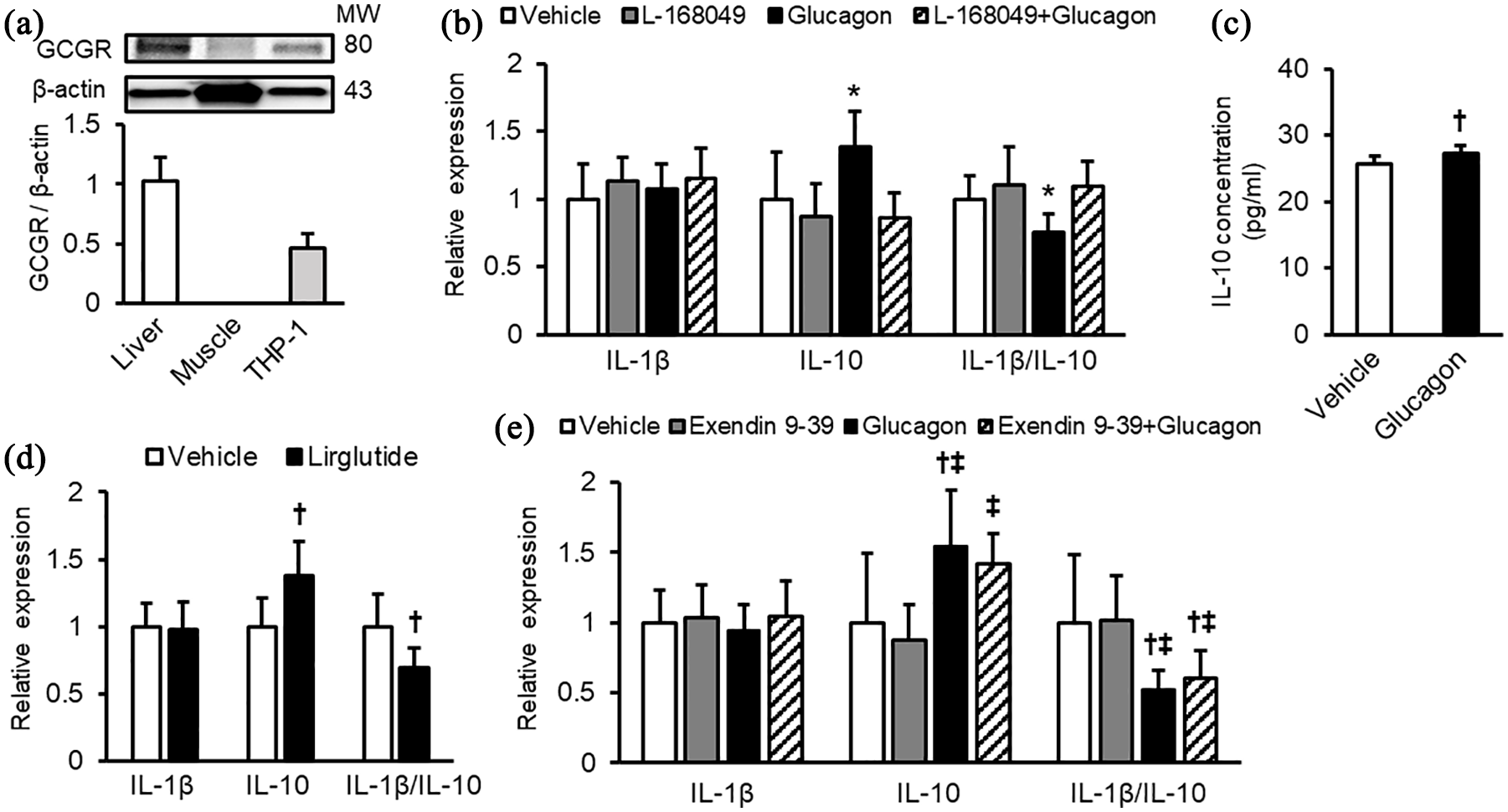
Anti-inflammatory effects of glucagon on cultured human monocytic THP-1 cells. (a) Protein levels of glucagon receptor (GCGR) in THP-1 cells. Liver and soleus muscle were used as positive and negative controls, respectively. Upper panels show representative immunoblot images. Protein levels were normalized by internal control β-actin levels and shown as relative expression to the livers. n = 3 per group. MW, molecular weight. (b) Effects of glucagon on gene expression levels in the absence and presence of the glucagon receptor antagonist, L-168049. (c) Effects of glucagon on IL-10 production by THP-1 cells. (d) Effects of liraglutide on gene expression levels. (e) Effects of glucagon on gene expression levels in the absence and presence of the GLP-1 receptor antagonist, exendin (9-39) amide. Gene expression levels of target molecules were normalized with those of the housekeeping gene 18S ribosomal RNA, and the data were shown as relative levels to the controls. n = 6–11 per group.
Discussion
Type 2 diabetes is one of the representative risk factors for atherosclerotic cardiovascular disease.8,9 Several types of oral hypoglycemic agents are shown to affect circulating glucagon levels.4,5 However, as far as we know, there is no study to examine the effects of glucagon in atherosclerosis of animal models or humans. In this study, we demonstrated for the first time that (1) a continuous infusion of glucagon attenuated the progression of atherosclerotic plagues and volumes in aortic sinuses and thoracic to abdominal aortae of ApoE−/− mice, an animal model of accelerated human atherosclerosis 10 and (2) the degree of plaque formations in both lesions were inversely correlated with plasma glucagon levels after 4-week interventions. Furthermore, we found here that collagen content and macrophage accumulation within the plaques of aortic sinuses were attenuated in glucagon-treated mice in association with the decreased ratio of Il-1β to Il-10 gene. Since glucagon infusion did not affect metabolic parameters or food intake except for modest decrease in fasting plasma glucose in high glucagon group, it is unlikely that the effects of glucagon on plasma glucose, lipid parameters and food intake could mainly contribute to atheroprotection in our animal models. Therefore, our present study suggests that glucagon might exert atheroprotective effects in mice partly via anti-inflammatory property.
In this study, we found that fasting plasma glucagon levels were increased by glucagon infusion from approximately 4.5 pmol/l in control mice to 8.0 and 18.5 pmol/l in the low-dose and high-dose glucagon-infused mice, respectively. A previous clinical study using the same glucagon ELISA kit as ours reported that fasting plasma glucagon levels were approximately 7.6 pmol/l in healthy young men and 18.1 pmol/l in obese type 2 diabetic men. 15 Therefore, physiological levels of glucagon may have atheroprotective property in vivo.
Chronic inflammation plays a central role in the instability of atherosclerotic plaques, where an imbalance between pro-inflammatory and anti-inflammatory cytokine productions from immune cells, such as macrophages are observed. 16 Among various cytokines, IL-1β and IL-10 are one of the representative pro-inflammatory and anti-inflammatory cytokines involved in atherosclerotic plaque instability, respectively.17–21 Indeed, a recent randomized double-blind clinical trial of canakinumab, a monoclonal antibody targeting IL-1β revealed that subcutaneous administration of 150 mg canakinumab every 3 months significantly inhibited major adverse cardiovascular events in patients with a history of myocardial infarction and a high-sensitivity C-reactive protein equal to or more than 2 mg/dl. 17 Furthermore, there is a growing body of evidence to suggest the atheroprotective role of IL-10.18–21 IL-10 transgenic mice exhibited less atherosclerotic lesions compared with wild-type mice, whereas IL-10-deficient mice was susceptible to atherosclerosis.18,19 Expression of IL-10, an anti-inflammatory cytokine mainly produced by macrophages and T cells are decreased within the human atherosclerotic plagues. 18 In addition, decreased IL-10 levels are associated with the increased intima media thickness of carotid arteries, 20 and increased serum levels of IL-10 could predict a more favorable prognosis in patients with acute coronary syndromes. 21 Therefore, we measured here gene expression levels of Il-1β and Il-10 in aortae and their ratio in order to evaluate the overall effects of glucagon on vascular inflammation within the atherosclerotic plagues. We found that glucagon infusion significantly decreased the ratio of Il-1β to Il-10. Furthermore, circulating IL-10 levels were elevated to 1.5- to 2.5-fold in glucagon group compared with vehicle group; IL-10 levels in high-dose glucagon group were significantly higher than those of vehicle group, whereas IL-1β in the blood were below the detectable limit of ELISA. Given that the increased ratio of IL-1β to IL-10 could contribute to the plaque instability partly by reducing the collagen content within the atherosclerosis via induction of matrix metalloproteinase activity, 16 although expression of just two factors may not adequately define the complex process of inflammation, glucagon may play a role in atherosclerotic plaque stability at least partly via the suppression of vascular inflammation.
In the present study, we found that glucagon receptor was actually expressed in human monocytic THP-1 cells. Moreover, glucagon was found to increase gene expression levels of IL-10 and resultantly decrease the ratio of IL-1β to IL-10, which was completely prevented by a glucagon receptor antagonist. Consistently, glucagon modestly, but significantly stimulated the IL-10 production released from THP-1 cells. Furthermore, we also examined the possibility whether glucagon at the applied dose (1 nM) could cross-react with GLP-1 receptors.22,23 We found that an antagonist for GLP-1 receptor did not affect gene expression levels of IL-1β or IL-10 in THP-1 cells, irrespective of the presence or absence of glucagon. Therefore, our present findings suggest that glucagon could work as an anti-inflammatory agent in THP-1 cells through the interaction with glucagon receptor. Although the underlying mechanisms how glucagon exerted anti-inflammatory effects on THP-1 cells remain unclear, glucagon has been shown to bind to glucagon receptor and subsequently exert various biological actions via a second messenger cyclic adenosine monophosphate (cAMP).1–3 Previous studies have shown that the cAMP-elevating agents exert anti-inflammatory effects on macrophages, such as induction of IL-10.24,25 Indeed, a GLP-1 receptor agonist liraglutide, which can increase intracellular cAMP levels, 26 mimicked the anti-inflammatory actions of glucagon in THP-1 cells. Therefore, glucagon and its receptor axis may decrease the ratio of IL-1β to IL-10 in macrophages, which could contribute to suppress the atherosclerotic plaque stability in ApoE−/− mice.
Conclusion
Our present findings suggest that glucagon may attenuate the progression of atherosclerotic plaques in ApoE−/− mice partly through its anti-inflammatory property.
Footnotes
Authors’ contributions
N.O. and H.K. performed experiments, analyzed data, interpreted results of experiments, drafted, and revised manuscript. Y.M. conceived and designed research, performed experiments, analyzed data, interpreted results of experiments, prepared figures, drafted manuscript, edited, and revised manuscript. T.S. performed experiments, analyzed data, interpreted results of experiments, and revised manuscript. M.H., M.T., H.Y., M.O., T.F., T.H., T.M., S.Y. interpreted results of experiments, and edited and revised manuscript. All authors have approved the final version of the manuscript. Y.M. is the guarantors of this work and are responsible for its integrity.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: S.Y. received honoraria such as lecture fees from Nippon Boehringer Ingelheim Co., Ltd., Sanofi K.K., Eli Lilly Japan K.K., and Ono Pharmaceutical Co., Ltd.