
Abstract
Peripheral artery disease is a severe complication of diabetes. We have reported that the deletion of angiotensin type 2 receptor in diabetic mice promoted vascular angiogenesis in the ischaemic muscle 4 weeks following ischaemia. However, the angiotensin type 2 receptor deletion beneficial effects occurred 2 weeks post surgery suggesting that angiotensin type 2 receptor may regulate other pro-angiogenic signalling pathways during the early phases of ischaemia. Nondiabetic and diabetic angiotensin type 2 receptor–deficient mice (Agtr2−/Y) underwent femoral artery ligation after 2 months of diabetes. Blood perfusion was measured every week up to 2 weeks post surgery. Expression of vascular endothelial growth factor, vascular endothelial growth factor receptor and endothelial nitric oxide synthase expression and activity were evaluated. Blood flow reperfusion in the ischaemic muscle of diabetic Agtr2+/Y mice was recovered at 35% as compared to a 68% recovery in diabetic Agtr2−/Y mice. The expression of vascular endothelial growth factor and its receptors was diminished in diabetic Agtr2+/Y mice, an observation not seen in diabetic Agtr2−/Y mice. Interestingly, Agtr2−/Y mice were protected from diabetes-induced glutathionylation, nitration and decreased endothelial nitric oxide synthase expression, which correlated with reduced endothelial cell death and enhanced vascular density in diabetic ischaemic muscle. In conclusion, our results suggest that the deletion of angiotensin type 2 receptor promotes blood flow reperfusion in diabetes by favouring endothelial cell survival and function.
Keywords
Introduction
Diabetes is associated with a high statistical risk of all-cause mortality and morbidity. The global prevalence of diabetes is estimated at 9% and diabetes causes approximately 1.5 million deaths per year. 1 Vascular complications of diabetes involve every tissue where there is terminal circulation as in the retina, kidney, heart and lower limbs. Reducing the cardiovascular risk in diabetic patients becomes a crucial research goal with the objective of preventing patients from atherosclerotic complications, endothelial dysfunction and peripheral artery disease (PAD). PAD is characterized by dysfunctional arterial vessel remodelling that leads to critical limb ischaemia, limb loss and even death. 1 Wound healing processes and ischaemic conditions require a highly regulated collateral vessel formation capability. Vascular endothelial growth factor (VEGF) has been shown to orchestrate the formation of new blood vessels during development and adulthood mainly by its tyrosine kinase receptor (VEGFR2). 2 This process is impaired by hyperglycaemia, which leads to a reduction in neovascularization and is correlated with a lower VEGF production in muscle tissue of diabetic animal models even under ischaemic stress.3,4 Beneficial effects of exogenous administration of VEGF reported in diabetic animal models have not improved neovascularization in humans in which the underlying explanation of this failure remains unclear. 5 Our group has previously reported that protein kinase C delta (PKC-δ) is up-regulated by hyperglycaemia in the ischaemic muscle which in turn reduced VEGF receptor activity due to enhanced SH2 domain containing phosphatase 1 (SHP-1) expression. 6
The renin angiotensin system (RAS) has been described as an important pathway involved in ischaemia-induced neovascularization by participating in endothelial and vascular smooth muscle cell proliferation and migration. However, it also increases oxidative stress, insulin resistance and the risk of diabetes. 7 Interestingly, in clinical trials, the angiotensin-converting enzyme inhibitors (ACEI) have demonstrated improvement in vascular outcomes in pathological conditions such as diabetes. 8 However, ACEI treatment raised the risk of developing diabetic foot ulcer and low extremity amputations. 9 The angiotensin type 1 receptor (AT1R) is the widely expressed angiotensin receptors in vascular tissue. Much less abundant, the angiotensin type 2 receptor (AT2R), has been shown to counteract AT1R effects. In previous reports, AT2R caused inhibition of AT1R angiogenic responses. 10 AT2R activation can raise the expression of pro-apoptotic protein BAX in cultured smooth muscle cells 11 as well as reduce VEGF-induced endothelial cell migration and tubule formation. 10 However, reports have indicated that AT2R may have favourable effects in myocardial hypertrophy and attenuated atherosclerosis through kinin/nitric oxide-dependent mechanisms. 12 In the context of diabetes, our group has recently published that activation of AT2R prevented VEGF actions on endothelial cell proliferation and migration. 13 This inhibition was associated with down regulation of ischaemia-induced angiogenesis caused by elevated expression of SHP-1. Interaction between AT2R/SHP-1 and VEGF signalling pathway has been demonstrated 4 weeks post ligation of the femoral artery. However, blood flow measurements indicated that reperfusion was almost completely recovered 2 weeks after surgery in diabetic AT2R-deficient mice. Therefore, we have conducted additional studies to investigate early mechanisms responsible for blood flow reperfusion improvement associated with AT2R deletion in diabetes.
Methods
Reagents and antibodies
Primary antibodies for immunoblot analyses were purchased from commercial sources, including actin (horseradish peroxidase (HRP); I-19), nitric oxide synthase (NOS) 3 (C-20), VEGF (147) antibodies from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA); p-eNOS (ser1177) antibodies from Cell Signaling (Danvers, MA, USA); anti-smooth muscle α-actin, mouse anti-3nitrotyrosine (39B6) and mouse anti-glutathione (D8) from Abcam (Cambridge, MA, USA); CD31 from BD Bioscience (San Jose, CA, USA); rabbit anti-3nitrotyrosine from Invitrogen (Waltham, MA, USA); rabbit and mouse peroxidase-conjugated secondary antibody also from Cell Signaling. Secondary antibodies Alexa-594–conjugated anti-rat IgG, Alexa-488–conjugated anti-rabbit IgG, Rhodamine Red-X anti-rabbit IgG and Fluorescein FITC anti-mouse IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). CGP 42112A was purchased from Bachem (Torrance, CA, USA), GKT136901 from Millipore (Etobicoke, ON, USA) and N-acetyl-cystein from Sigma (St. Louis, MO, USA).
Animal and experimental design
C57BL/6J (Ins2 +/+) and diabetic heterozygous male Ins2+/C96Y (diabetes mellitus (DM); Akita) mice were purchased from The Jackson Laboratory and bred with the Agtr2−/Y mice in our animal facility for seven generations as described previously. 13 Each group included 12 mice. Throughout the entire study, animals were provided with free access to water and standard rodent chow (Harlan Teklad). All experiments were performed according to the Canadian Council of Animal Care and Institutional Guidelines and were approved by the Animal Care and Use Committees of the University of Sherbrooke, according to National Institutes of Health (NIH) guidelines.
Hindlimb ischaemia model
We have measured blood flow reperfusion in nondiabetic (NDM; Ins2 +/+) and 4-month-old diabetic (DM; Ins2 +/C96Y) Agtr2 +/Y and Agtr2−/Y mice as described previously. 13 Briefly, mice were anaesthetized and the entire lower extremity was shaved. A small incision was made along the thigh all the way to the inguinal ligament and extended superiorly towards the mouse abdomen. The right femoral artery was isolated from the femoral nerve and vein, and ligated distally to the origin of the arteria profunda femoris. The incision was closed by interrupted 5-0 sutures (Syneture). All animals were followed out to 14 days post surgery and subsequent experiments were performed on day 14 postoperative ischaemic tissue.
Laser Doppler perfusion imaging and physical examination
Lower limb blood flow was assessed using a laser Doppler perfusion imaging (PIMIII) system (Perimed Inc, Ardmore, PA, USA). Consecutive perfusion measurements were acquired by scanning the region of interest (hindlimb and foot) of anaesthetized animals. Measurements were performed pre- and post-artery ligation, and additionally on postoperative days 7 and 14. To account for variables that may influence blood flow temporally, the results at any given time were expressed as a ratio against simultaneously obtained perfusion measurements of the right (ligated) and left (non-ligated) limb of 12 mice per group.
Histopathology
Right abductor muscles from nondiabetic and diabetic Agtr2 +/Y and Agtr2−/Y mice were harvested for pathological examination and sections were fixed in 4% paraformaldehyde (Sigma–Aldrich) for 18 h and then transferred to 70% ethanol. Paraformaldehyde-fixed tissue was embedded in paraffin and 4-µm sections were stained with hematoxylin and eosin (Sigma) as previously described. 13 The entire tissue sample of the muscle fibre structure of six mice per group was visualized under light microscope on a Nikon eclipse TI.
Immunofluorescence and TUNEL assay
The ischaemic abductor muscles were blocked with 10% goat serum for 1 h and were exposed to primary antibodies (CD31 (1:50), α-smooth muscle actin, 1:200) and 3-nitrotyrosine (1:200) overnight, followed by incubation with secondary antibodies (Alexa-488–conjugated anti-rabbit IgG, Alexa-594–conjugated anti-rat, Rhodamine Red-X anti-rabbit IgG (1:400) and Fluorescein FITC anti-rat IgG (1:200)). The number of vessels smaller than 35 µm (measured by the vessel lumen diameter) was counted in cross section of the entire ischaemic muscle sample per mm2. Apoptotic cells were detected using the TACS 2 Tdt-Fluor in situ apoptosis detection kit (Trevigen, Gaithersburg, MD, USA) according to the manufacturer’s instructions and as described previously. Briefly, sections were incubated with CD31 (1:50) overnight followed by incubation with secondary antibody Alexa-488–conjugated antibody. Proteinase K treatment was performed for 30 min at 37°C. Anti-streptavidin 647 was used to observe apoptotic cells. Images were captured on a Nikon eclipse Ti microscope; images of one experiment were taken at the same time under identical settings and similarly handled in Adobe Photoshop across all images. The quantification of nitrotyrosine and CD31 colocalization was performed by counting both the number of CD31 only and CD31/nitrotyrosine positive cells using the Image J software. The percentage of colocalization was then calculated dividing the number of CD31/nitrotyrosine positive cells by the total number of CD31 positive cells. All quantifications were carried out in a blinded manner. Six (immunofluoprescence of CD31 and α-smooth muscle actin) and seven (terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay) mice per group were used.
Coimmunoprecipitation and immunoblot analysis
The ischaemic abductor muscles were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mmol/L Tris (pH 6.8), 150 mmol/L NaCl, 0.1% sodium dodecyl sulphate (SDS), 1% triton, 0.5% sodium deoxycholate and 2 mmol/L ethylenediaminetetraacetic acid (EDTA)) containing protease inhibitors (1 mmol/L phenylmethylsulfonyl fluoride, 2 µg/mL aprotinin, 10 µg/mL leupeptin, 1 mmol/L Na3VO4; Sigma). Protein amount was measured with DC kit (Bio-Rad, Hercules, CA, USA.). Coimmunoprecipitation assays were performed as described previously. 14 Briefly, 500 μg of lysate proteins was incubated for 90 min with 1 μg of antibodies specific against endothelial nitric oxide synthase (eNOS). Then 35 μL of precleaned A/G protein-coated agarose beads were added to the mix and rotated at 4°C for 30 min. Beads were then spun-down and washed to isolate the protein complex containing the protein of interest and followed by the addition of 25 μL of 2× Laemmli (without Bêta-mercaptoethanol). For immunoblot analysis, the lysates (20–50 µg protein) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membrane, and blocked with 5% skim milk. Primary antibody was incubated overnight in 5% skim milk (eNOS 1:1000 and 3-nitrotyrosine 1:2000) or in 5% bovine serum albumin (BSA) (p-eNOS 1:500 and glutathione 1:1000). Antigens were detected using anti-rabbit HRP–conjugated antibody 1:10,000 or anti-mouse HRP–conjugated antibody 1:10,000 or 1:5000 and detected with Luminata forte western HRP substrate (Millipore). Protein content quantification was performed using computer-assisted densitometry ImageLab imaging software, Chemidoc, BioRad. In total, 6–8 mice per group were used.
Real time PCR analysis
Real-time polymerase chain reaction (PCR) (Sybr Green) was performed to evaluate mRNA expressions of VEGF, Flk-1/KDR, platelet-derived growth factor (PDGF) and platelet-derived growth factor receptor (PDGFR) of ischaemic limbs. Total RNA was extracted from the ischaemic abductor muscles with TRI-REAGENT, as described by the manufacturer and RNeasy mini kit (Qiagen, Valencia, CA, USA). The RNA was treated with deoxyribonuclease I (DNase I; Invitrogen) to remove any genomic DNA contamination. Approximately 1 µg RNA was used to generate cDNA using SuperScript III reverse transcriptase and random hexamers (Invitrogen) at 50°C for 60 min. PCR primers are listed in Supplementary Table 1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was used for normalization. PCR products were gel purified, subcloned using QIA quick PCR Purification kit (Qiagen), and sequenced in both directions to confirm identity. In total, 6 mice per group were used.
Cell culture
Endothelial cells were extracted from bovine aorta as described previously. 13 Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) 2.5% foetal bovine serum (FBS), 1% P/S until they reached 80% confluence. Cells were then treated in DMEM 0.1% FBS containing normal glucose (NG; 5.6 mmol/L + 19.4 mmol/L mannitol to adjust osmotic pressure) levels for 24 h, with or without AT2 antagonist (CGP 42112A; 10 nmol/L), Nox1/4 inhibitor (GKT136901; 200 nm) and or N-acetyl-cystein (500 µm).
Statistical analyses
The data were shown as mean ± SEM for each group. Statistical analysis was performed by unpaired t test or by one-way analysis of variance (ANOVA) followed by Tukey’s test correction for multiple comparisons. Data in each group were checked for normal distribution using D’Agostino and Pearson normality test based on alpha = 0.05. All results were considered statistically significant at p < 0.05.
Results
Deletion of AT2 receptor improves reperfusion and reduces vascular cell death after ischaemia in diabetic limbs
Nondiabetic and diabetic Agtr2+/Y and Agtr2−/Y mice underwent right femoral artery ligation. The deletion of the AT2 receptor had no effect on body weight and fasting glucose levels (Table 1). Blood reperfusion was assessed using the laser Doppler imaging system (Figure 1(a)). Results showed a 48% decrease of blood flow reperfusion in diabetic Agtr2+/Y (35% blood flow recovery) compared with nondiabetic Agtr2+/Y mice (67% blood flow recovery; p = 0.0025). In contrast, reperfusion of blood supply to the limb following ischaemia of diabetic Agtr2−/Y mice was significantly improved compared with diabetic Agtr2+/Y mice (p = 0.0023), reaching 68% recovery, a level that was similar to nondiabetic Agtr2+/Y mice (Figure 1(b)). More importantly, diabetic Agtr2+/Y mice displayed a 34% vascular density reduction as compared with nondiabetic Agtr2+/Y mice (p = 0.0027). In contrast, diabetic Agtr2−/Y mice displayed a significantly higher number of small vessels (smaller than 35 µm) and vascular density compared with diabetic Agtr2+/Y mice (Figure 1(c) and (d)).
Body weight and fasting glucose levels of Ins2+/+–Agtr2+/Y, Ins2+/C96Y–Agtr2+/Y, Ins2+/+–Agtr2–/Y and Ins2+/C96Y–Agtr2–/Y mice.
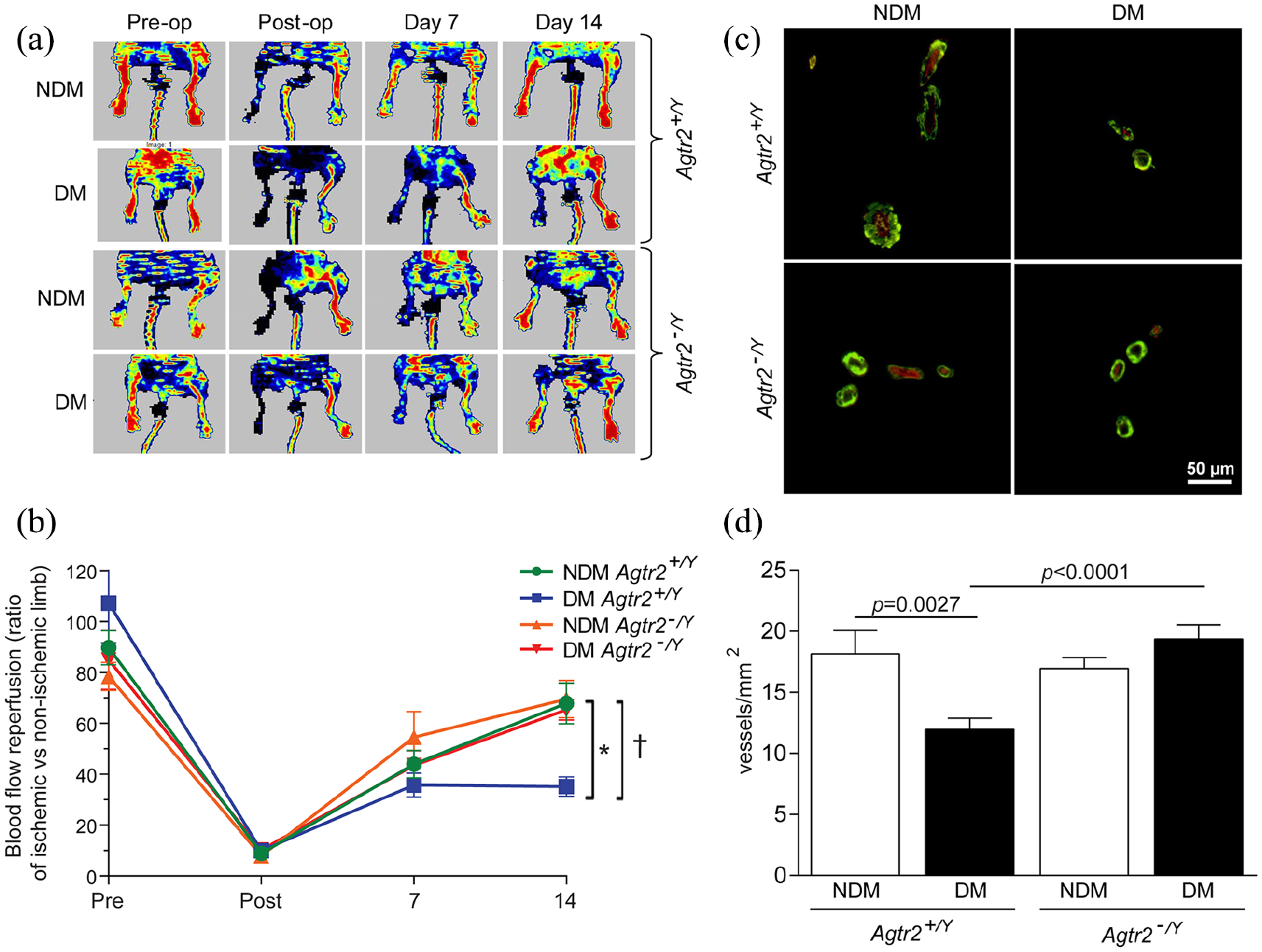
Blood flow reperfusion and vascular density are improved in diabetic Agtr2-/Y mice: (a) blood flow reperfusion images of nondiabetic and diabetic Agtr2+/Y and Agtr2−/Y mice before, after, and 2 weeks following femoral artery ligature, (b) quantification of the percentage of blood flow reperfusion using the laser Doppler imaging system, (c) immunofluorescence of α-smooth muscle actin (green) and endothelial cells (CD31-red) in the ischaemic abductor muscle of nondiabetic and diabetic Agtr2+/Y and Agtr2−/Y mice and (d) quantification of vascular density (number of vessels smaller than 35 µm). Results are shown as mean ± SEM of 12 mice per group (A and B) and 6 mice per group (C and D).*p = 0.0025 versus nondiabetic Agtr2+/Y, †p = 0.0023 versus diabetic Agtr2−/Y.
Vascular density is preserved in absence of AT2R in the ischaemic muscle of diabetic mice
Histology analysis showed a remarkable maintenance of the muscle structure and integrity after 2 weeks of ischaemia in diabetic Agtr2−/Y mice that was similar to its nondiabetic counterpart Agtr2−/Y mice, an observation that was not found in diabetic Agtr2+/Y mice (Figure 2(a)). Besides angiogenesis, another potential mechanism that could explain the poor blood flow reperfusion in diabetic mice is due to vascular cell apoptosis during ischaemia. Therefore, we performed immunofluorescence analysis of endothelial cells that underwent cell death using TUNEL assay in ischaemic muscle cross-sections of our four groups of mice. As expected, diabetic Agtr2+/Y mice showed significantly elevated endothelial cell apoptosis as compared to nondiabetic Agtr2+/Y mice (p = 0.0219). The absence of AT2R protected endothelial cells from apoptosis in a diabetic state (Figure 2(b) and (c)).
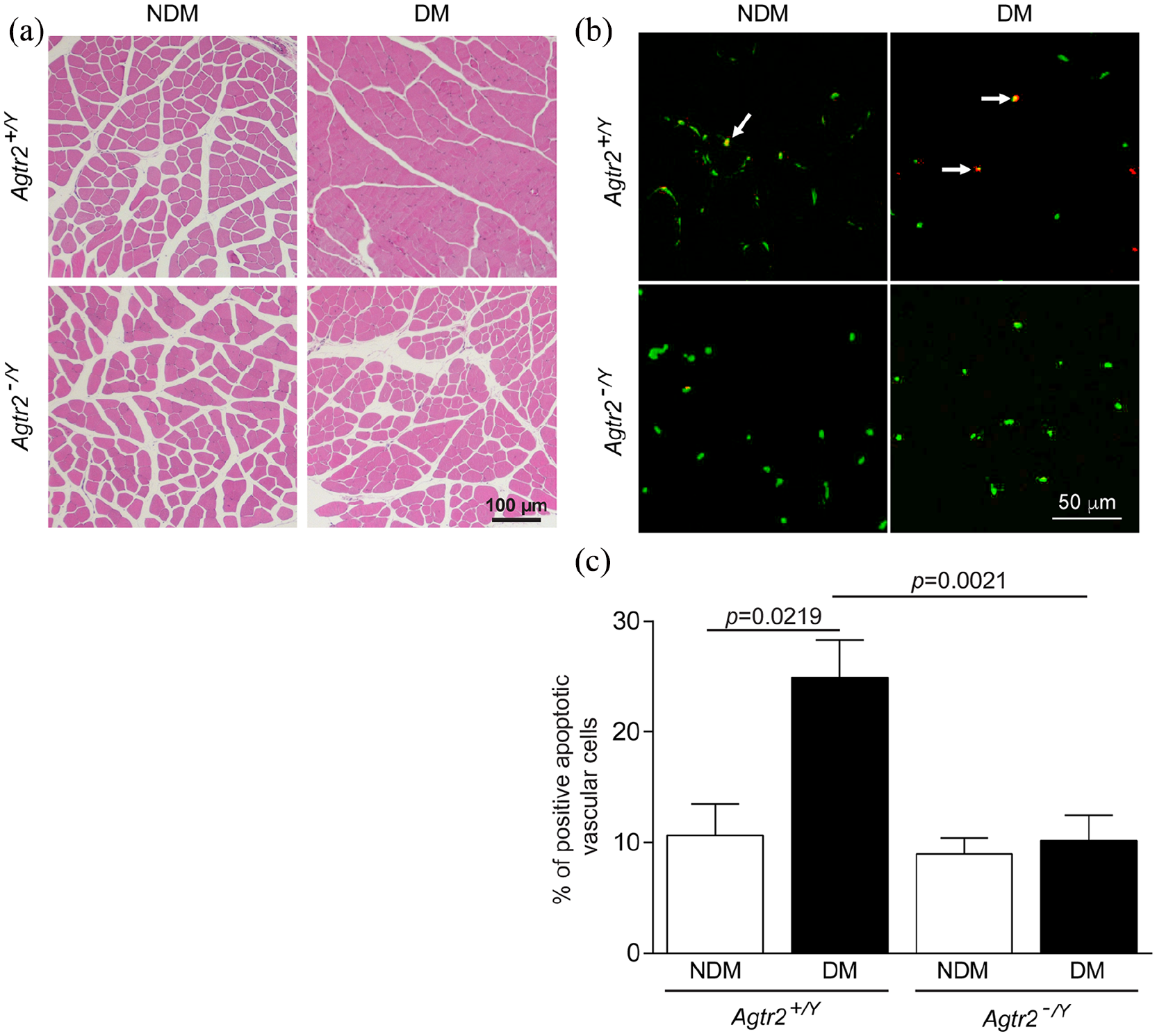
Ablation of AT2R in diabetes preserved muscle structure integrity and vascular apoptosis: (a) structural analysis of the ischaemic muscle after hematoxylin and eosin staining of nondiabetic and diabetic Agtr2+/Y and Agtr2−/Y mice, (b) immunofluorescence of vascular endothelial cells (CD31-green) and positive apoptotic cells (red) and (c) quantification of the percentage of vascular endothelial apoptotic cells (white arrows) per mm2 was determined by counting positive apoptotic cells which colocalized with CD31 per total count of CD31 positive cells. Results are shown as mean ± SEM of ischaemic sections of 6 mice per group (A) and 7 mice per group (B and C).
Deletion of AT2R promotes pro-angiogenic growth factors expression
Since hypoxia is the major component of angiogenesis induction and in order to explain how the deletion of AT2R prevented vessel density and blood flow reperfusion impairment observed in diabetic mice, mRNA expression of critical angiogenic factors was measured. As we previously reported, diabetes decreased VEGF and Flk-1 mRNA expression in the ischaemic muscle. The ablation of AT2R completely and significantly prevented the decrease of VEGF and Flk-1 mRNA expression observed in diabetic mice (Figure 3(a) and (b)). In contrast to our data after 4 weeks of femoral artery ligation, we did not detect any change of PDGF or PDGFR mRNA expression (Figure 3(c) and (d)). Since eNOS activity is linked to VEGF expression, eNOS protein expression and phosphorylation were measured by immunoblot. As shown in Figure 4(a) and (b), both the phosphorylation and protein expression of eNOS were significantly reduced by 71% (p = 0.0476) and 60% (p = 0.0183), respectively, and correlated with decreased VEGF expression (Figure 4(c); p = 0.0040) in the ischaemic muscle of diabetic Agtr2+/Y as compared to nondiabetic Agtr2+/Y. Interestingly, the absence of AT2R prevented the loss of eNOS expression (Figure 4(a); p = 0.0037) and phosphorylation (Figure 4(b); p = 0.0132) as well as VEGF expression (Figure 4(c); p = 0.0114) in the ischaemic muscle of diabetic mice.
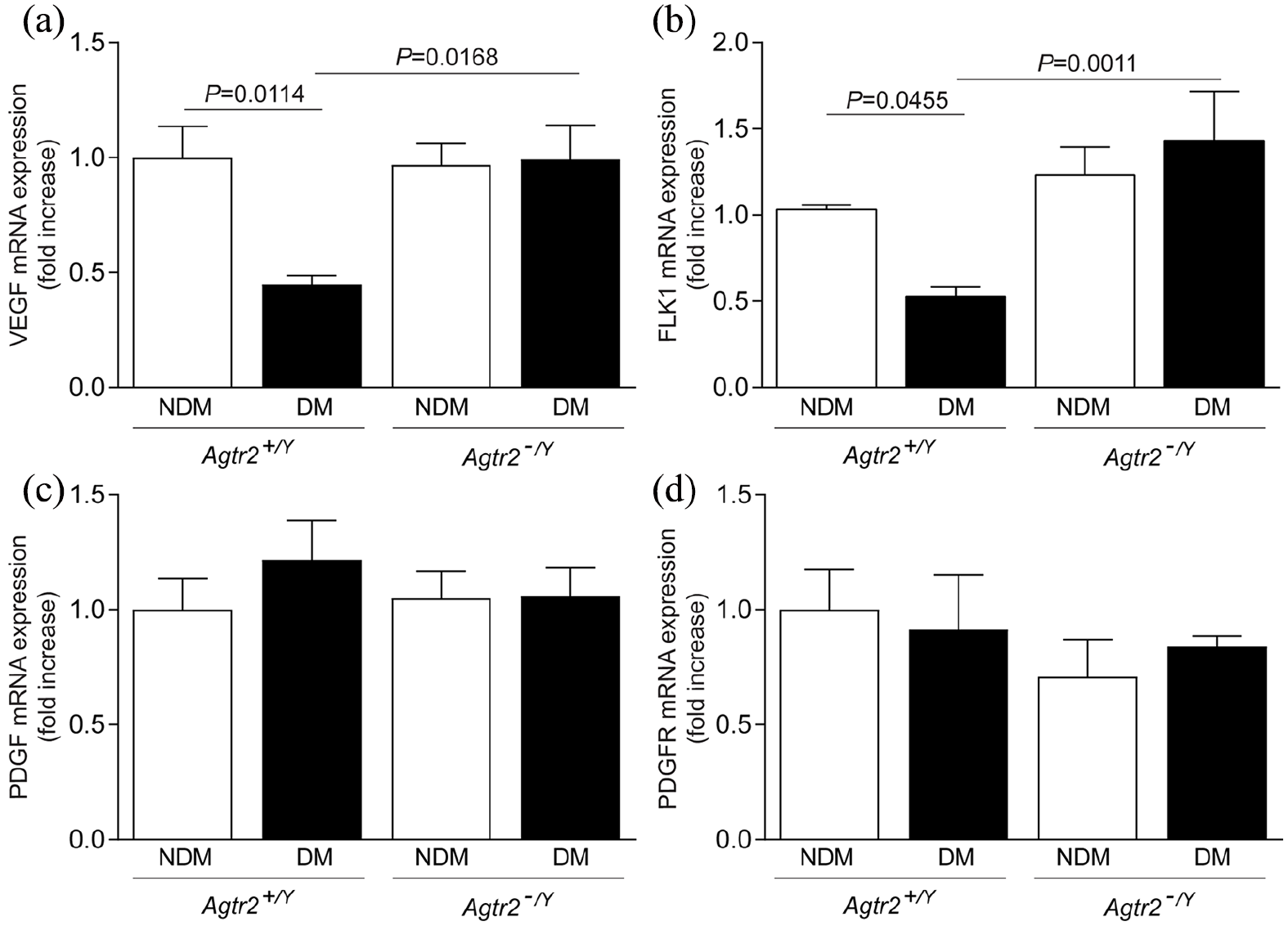
VEGF and VEGFR, but not PGDF mRNA expression were enhanced in the muscle of diabetic Agtr2−/Y mice: (a) VEGF, (b) VEGFR Flk-1, (c) PDGF and (d) PDGFR expression were measured in the abductor ischaemic muscle of nondiabetic and diabetic Agtr2+/Y and Agtr2−/Y mice. Results are shown as mean ± SEM of 6–8 mice per group.
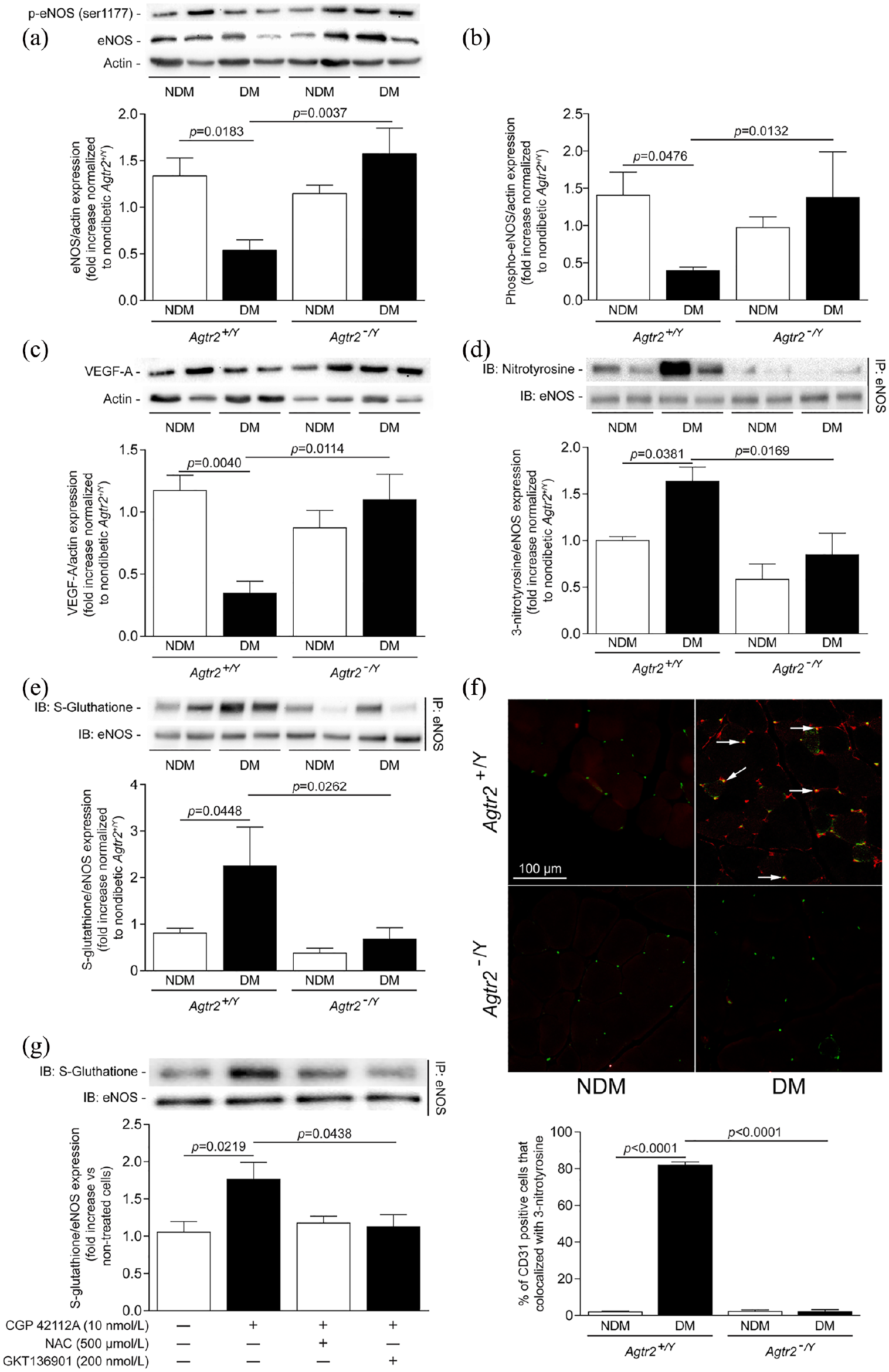
AT2R-deficient mice were protected from diabetes-induced reduction of eNOS expression, and increased glutathionylation and nitration of eNOS. Immunoblot analysis of (a) eNOS, (b) phospho-eNOS, (c) VEGF, (d) nitrotyrosine and (e) S-glutathione expression following the immunoprecititation of eNOS, (f) immunofluorescence of endothelial cell marker CD31 in green and 3-nitrotyrosine expression (in red) in the abductor ischaemic muscle of nondiabetic and diabetic Agtr2+/Y and Agtr2−/Y mice and (g) immunoblot analysis of S-glutathione following the immunoprecipitation of eNOS of endothelial cells treated with or without CGP 42112A (10 nM), Nox inhibitor (200 nM) and NAC (500 µM). Results are shown as mean ± SEM of 6 mice per group (A–E, G) and 8 mice per group (F).
Deletion of AT2 receptor prevents S-glutathionylation and 3-nitrotyrosination of eNOS caused by diabetes
Since diabetes is associated with increased oxidative stress that commonly generate the uncoupling of eNOS, 15 we have performed additional experiments to determine if AT2R could be involved in diabetes-induced eNOS glutathionylation and nitration, two markers associated with eNOS uncoupling. The expression of tyrosine nitration (Figure 4(d)) and S-glutationylation (Figure 4(e)) of eNOS was significantly enhanced in muscle of diabetic Agtr2+/Y mice compared to nondiabetic Agtr2+/Y mice, which was attenuated in diabetic Agtr2−/Y mice (Figure 4(d) and (e)). In addition, the nitrotyrosination signal was predominant in endothelial cells (marker by CD31) of ischaemic muscle of diabetic Agtr2+/Ymice compared to nondiabetic Agtr2+/Y and diabetic Agtr2−/Y mice (Figure 4(f)). Interestingly, the S-glutathionylation of eNOS can be induced by an agonist of AT2R (CGP 42112A) in cultured endothelial cells, which can be prevented by both antioxidant (NAC) and Nox1/4 inhibitor (GKT136901) (Figure 4(g)).
Discussion
Angiogenesis, defined as sprouting of new vessels from pre-existing vasculature, is a physiological response to ischaemia. One of the main angiogenic factors implicated in this process is VEGF which activates a series of pathways including Akt and eNOS. 16 VEGF triggers neovascularization in nondiabetic patients with limb ischaemia as well as in some microvascular complication of diabetes such as retinopathy and nephropathy. 17 However, despite that hypoxia and oxidative stress, the major inducers of VEGF production, are elevated in diabetes, decreased VEGF expression in the peripheral limbs and nerves of diabetic animals have been reported by us and other laboratories.3,18 We have recently reported that diabetes-induced AT2R activation led to increased SHP-1 activity and reduced VEGF actions in the ischaemic muscle after 4 weeks of femoral artery ligation. Since we observed that blood flow reperfusion was almost completely restored after 2 weeks following ischaemia, 13 this study investigated the effect of AT2R ablation on neovascularization in the ischaemic hindlimb in diabetic mice after 14 days post surgery. Deletion of AT2R significantly prevented endothelial cell death in the ischaemic muscle of diabetic mice as compared to diabetic Agtr2+/Y mice. In addition, the density of small vessels was enhanced in diabetic Agtr2−/Y mice, which was associated with preservation of VEGF, Flk-1 and eNOS expression in the ischaemic limb. Measuring vessels smaller than 35 µm allowed us to evaluate the number of capillaries (5–10 µm) and arterioles (average diameter of 30 µm), which reflects in part the formation of newly small vessels during the angiogenesis process.
Alteration of the angiogenesis response by hyperglycemia depends on the targeted tissue. The retina and the myocardium react very differently to hypoxia. Expression of VEGF is decreased in the myocardium and peripheral vessels but elevated in the retina of proliferative retinopathy of diabetic animals and patients.19,20 One potential mechanism by which diabetes generates opposite angiogenic responses in several tissues could be due to local RAS receptor activation. As reported previously, neovascularization was diminished in the hindlimb when AT2R was activated, and was associated with inhibition of VEGF expression and fewer collateral vessel formation. 21 Meanwhile, in other tissues such as the heart, Munk et al. 22 demonstrated that angiotensin II-induced sprouting was absent in Agtr2−/Y suggesting that AT2R is necessary for the angiogenesis process during cardiac repair. Interestingly, in this study, the authors showed that deletion of AT2R did not affect VEGF actions but rather involved the bradykinin receptor signalling pathway. In our study, apart from the angiogenesis response, we found that the vascular endothelial cell apoptosis index was higher in Agtr2+/Y diabetic mice compared with Agtr2−/Y diabetic mice. These data correlated with decreased capillary density and reduced VEGF and VEGF receptor mRNA expression. A previous study indicated that the angiogenic properties of Ang II to enhance VEGF and VEGFR expression could be due to the activation of AT1R. 23 Therefore, in absence of AT2R, it is possible that the local RAS activity could be mediated via the AT1R to promote vessel formation in the ischaemic muscle.
An ample number of publications have established the importance of VEGF-mediated NO production, an essential mechanism for endothelial cell migration and tubule formation. Previous in vitro experiments demonstrated that stimulation of AT2R with CGP 42112A prevented VEGF-induced Akt and eNOS phosphorylation. 10 Our previous study showed that the phosphorylation of the VEGFR2, and subsequently Akt was reduced in diabetic animals and completely prevented in the muscle of Agtr2−/Y mice 4 weeks post surgery. 13 This study enhanced our knowledge that AT2R activation can also affect eNOS in vivo by decreasing both the phosphorylation and expression of eNOS. Numerous studies reported compelling evidence that eNOS uncoupling, which is characterized by the generation of superoxide instead of NO, has been viewed has an important mechanism of endothelial dysfunction in diabetes. 24 S-glutathionylation is a post-transcriptional modification characterized by the binding of a glutathione tripeptide to the protein through the formation of a disulfide bond with the protein thiol. Previously, Chen et al. 25 showed that eNOS S-glutathionylation in endothelial cells switched from NO production to superoxide production and this was associated with impaired endothelium-dependent vasodilation. Our data demonstrated an increased S-glutathionylation of eNOS in diabetic muscle and endothelial cells exposed to AT2 agonists, which was prevented by the absence of AT2R and by reducing oxidative stress (NAC and Nox1/4 inhibitor). Interestingly, another study has reported that eNOS S-glutathionylation is a key mediator of angiotensin II-induced endothelial dysfunction, an effect that was inhibited with the treatment of an ACE inhibitor. 26 In addition, elevated cellular tyrosine nitration has been linked to uncoupling of eNOS and reduction of flow-mediated dilation of coronary arterioles of diabetic patients. 27 Our results also indicated that tyrosine nitration of eNOS was increased in muscle of diabetic animals that was prevented in diabetic AT2R-deficient mice. Overall, these data suggest that activation of AT2R in the context of diabetes can lead to eNOS glutathionylation and nitration, and poor collateral vessel formation. However, our results are in contrast with previous rodent experiments which reported that direct activation of AT2R with a selective agonists (CGP 42112A and C21 compound) exerted beneficial effects of cardiac and brain microvascular function through an elevation of NO production and brain-derived neurotrophic factor, and suppression of the inflammatory processes.28,29 Another study showed that the overexpression of AT2R in a pro-atherosclerosis mouse model decreased lipid deposition in the aorta through reduced production of superoxide by the mononuclear leukocytes rather than a direct effect on the vascular oxidative stress. 12 Therefore, the positive or negative response to vascular remodelling mediated by AT2R activation could be influenced by the environment (nondiabetic vs diabetic) and cell type.
In addition to its effect on VEGF and eNOS actions, AT2R downregulation significantly reduced endothelial cell apoptosis induced by diabetes in the ischaemic muscle. Our data corroborate previous observation that indicated an elevation of Bcl-2, a cell survivor factor that can delay apoptotic processes induced by a variety of stimuli 30 . Furthermore, we cannot exclude possible beneficial effect of AT2R deletion on smooth muscle cells, which play an important role during the angiogenesis process. Previous reports showed that AT2R-mediated apoptosis and inhibition of AT1R-mediated proliferation of smooth muscle cells. 31 Therefore, it is possible that, combined to its positive effect on endothelial cell function preservation, inhibition of AT2R enhanced angiogenesis through smooth muscle cell growth.
In conclusion, AT2R deletion prevented endothelial cell apoptosis and restored endothelial cell function to promote angiogenesis in the ischaemic limb of diabetic mice. These results provided novel insights and new perspectives for prevention and treatment of peripheral artery disease complications in diabetic patients.
Supplemental Material
supplementary_table – Supplemental material for Ablation of angiotensin type 2 receptor prevents endothelial nitric oxide synthase glutathionylation and nitration in ischaemic abductor muscle of diabetic mice
Supplemental material, supplementary_table for Ablation of angiotensin type 2 receptor prevents endothelial nitric oxide synthase glutathionylation and nitration in ischaemic abductor muscle of diabetic mice by Stéphanie Robillard, Clément Mercier, Valérie Breton, Judith Paquin-Veillette, Andréanne Guay, Farah Lizotte and Pedro Geraldes in Diabetes & Vascular Disease Research
Footnotes
Acknowledgements
The authors gratefully acknowledge Anne Vezina (Department of Cell Anatomy, University of Sherbrooke) for her assistance with the histology technics. Dr Geraldes is the guarantor of this work, had full access to all the data, and takes full responsibility for the integrity of the data and the accuracy of data analysis.
Author contributions
S.R., C.M., V.B., J.P-V and F.L. performed experiments. A.G. performed animal care and researched data. S.R., F.L. and P.G. analysed the data and wrote the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by grants from the Canadian Institute of Health Research (PTJ159627 to P.G.). This work was performed at the CHUS research centre, funded by the ‘Fonds de Recherche du Québec – Santé’ (FRQ-S). Dr Geraldes is currently the holder of the Canada Research Chair in Vascular Complications of Diabetes.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.