
Abstract
While few dispute the existence of the metabolic syndrome as a clustering of factors indicative of poor metabolic health, its utility above that of its individual components in the clinical care of individual patients is questioned. This is likely a consequence of the failure of clinicians and scientists to agree on a unifying mechanism to explain the metabolic syndrome. Insulin resistance has most commonly been proposed for this role and is generally considered to be a root causative factor for not only metabolic syndrome but also for its associated conditions of non-alcoholic fatty liver disease (NAFLD), polycystic ovary syndrome (PCOS), obesity-related type 2 diabetes (T2D) and atherosclerotic cardiovascular disease (ASCVD). An alternative view, for which evidence is mounting, is that hyper-responsiveness of islet β-cells to a hostile environment, such as westernised lifestyle, is primary and that the resulting hyperinsulinaemia drives the other components of the metabolic syndrome. Importantly, within this new conceptual framework, insulin resistance, while always a biomarker and state of poor metabolic health, is not considered to be harmful, but a protective adaptive response of critical tissues including the myocardium against insulin-induced metabolic stress. This major shift in how metabolic syndrome can be considered puts insulin hypersecretion into position as the unifying mechanism. If shown to be correct, this new conceptual framework has major implications for the future prevention and management of the metabolic syndrome, including its associated conditions of NAFLD, PCOS, obesity-related T2D and ASCVD.
Keywords
Professor Gerald ‘Jerry’ Reaven, in his 1988 American Diabetes Association Banting Lecture titled ‘Role of insulin resistance in human disease’, showed strong associations between insulin resistance, hyperinsulinaemia, glucose intolerance, hypertriglyceridaemia, reduced high-density lipoprotein cholesterol and hypertension. 1 He termed the clustering of these factors ‘syndrome X’ and demonstrated links between this syndrome and increased risk of atherosclerotic cardiovascular disease (ASCVD). 1 Syndrome X, renamed ‘metabolic syndrome’ (MetS), has been expanded to include additional factors such as central or visceral adiposity, increased apolipoprotein B and small dense low-density lipoprotein (LDL) particles (proatherogenic), elevated plasma fibrinogen and plasminogen activator inhibitor (PAI)-1 (prothrombotic), increased C-reactive protein and inflammatory cytokines (systemic inflammation) and microalbuminuria.2,3 It is generally accepted that the clustered components of the MetS, including insulin resistance, contribute to the pathogenesis of conditions such as non-alcoholic fatty liver disease (NAFLD), polycystic ovary syndrome (PCOS), type 2 diabetes (T2D) and ASCVD.2–5 MetS has also been associated with increased risk for chronic kidney disease, cognitive impairment, obstructive sleep apnoea and chronic respiratory diseases.6–9 While the usefulness of a diagnosis of MetS over its individual components in predicting T2D and ASCVD has been questioned, MetS is now listed as a disease entity (E88.81) in the International Classification of Diseases – 10th Revision (ICD-10), avowing to the importance of Reaven’s contribution in bringing this clustering of factors involved in cardiometabolic diseases to the attention of clinicians and scientists.1,10
Insulin resistance: root cause of MetS and T2D or a protective adaptive response?
Ongoing controversy surrounding the MetS, in terms of its predictive value for particular diseases, is a consequence of the failure of metabolic scientists and clinicians to establish it as a precise condition or to provide a unifying mechanism to explain its clustering of factors, with insulin resistance and visceral adiposity being most commonly proposed.2,10 Reaven argued for insulin resistance as the unifying mechanism or primary causal factor and, supporting this view, the European Group for the Study of Insulin Resistance proposed ‘insulin resistance syndrome’ as an alternate name for MetS.1,11 Furthermore, the mainstream understanding of pathogenesis of T2D is that it develops as a consequence of failure of pancreatic islet β-cells to sustain the hyperinsulinaemia required to compensate for insulin resistance, giving insulin resistance a high-level causative role.12,13 Thus, within the current conceptual framework, insulin resistance is considered to be ‘harmful’ and the root cause of T2D and all the other conditions linked to the MetS; furthermore, it should be overcome at any cost.
An alternate view gaining momentum is that insulin resistance has a role in protecting critical tissues of the body from metabolic injury in situations of chronic nutrient excess.14–17 Its presence within the MetS, while indicative and a biomarker of poor metabolic health, does not mean insulin resistance has a causative role. Furthermore, if insulin resistance does have an adaptive protective role, attempts to override it in patient treatment have the potential to cause harm. Thus, we believe a shift is needed in the conceptual framework by which we understand insulin resistance and the aetiology of T2D and that this has implications on safe management of patients with MetS, T2D and related conditions.
Insulin sensitivity: adaptable to physiological demands
Physiological adaptability in insulin sensitivity is an important mechanism by which the body can regulate nutrient partitioning between tissues, necessitated by wide fluctuations in dietary intake and physical activity, and life events such as rapid pubertal growth, pregnancy, illness and ageing. For example, in response to short-term overfeeding, a rapid fall in insulin sensitivity occurs which allows diversion of nutrients from skeletal muscle to adipose tissue for storage, potentially important moving between situations of feast and famine.18,19 Pregnancy necessitates diversion of nutrients to the developing foetus and insulin resistance in the mother is a mechanism by which this is achieved. 20 Key to this discussion is the role of adaption in insulin sensitivity to a chronic nutrient oversupply, as occurs in westernised lifestyles. As discussed below, the development of insulin resistance in such situations could provide important protection to critical tissues such as the heart from nutrient overload and toxicity.14–17
Insulin resistance: a protective mechanism against nutrient-induced intracellular metabolic stress
We previously proposed that in response to chronic over-nutrition, tissues normally responsive to insulin for glucose uptake, such as the heart and skeletal muscle, protect themselves from nutrient-induced toxicity by becoming insulin resistant.14,15 Without this mechanism at times of nutrient surplus, or by overriding this protective insulin resistance with high-dose insulin therapy, these tissues will be damaged by nutrient overload, a process we have termed ‘insulin-induced metabolic stress’ (Figure 1).14,15
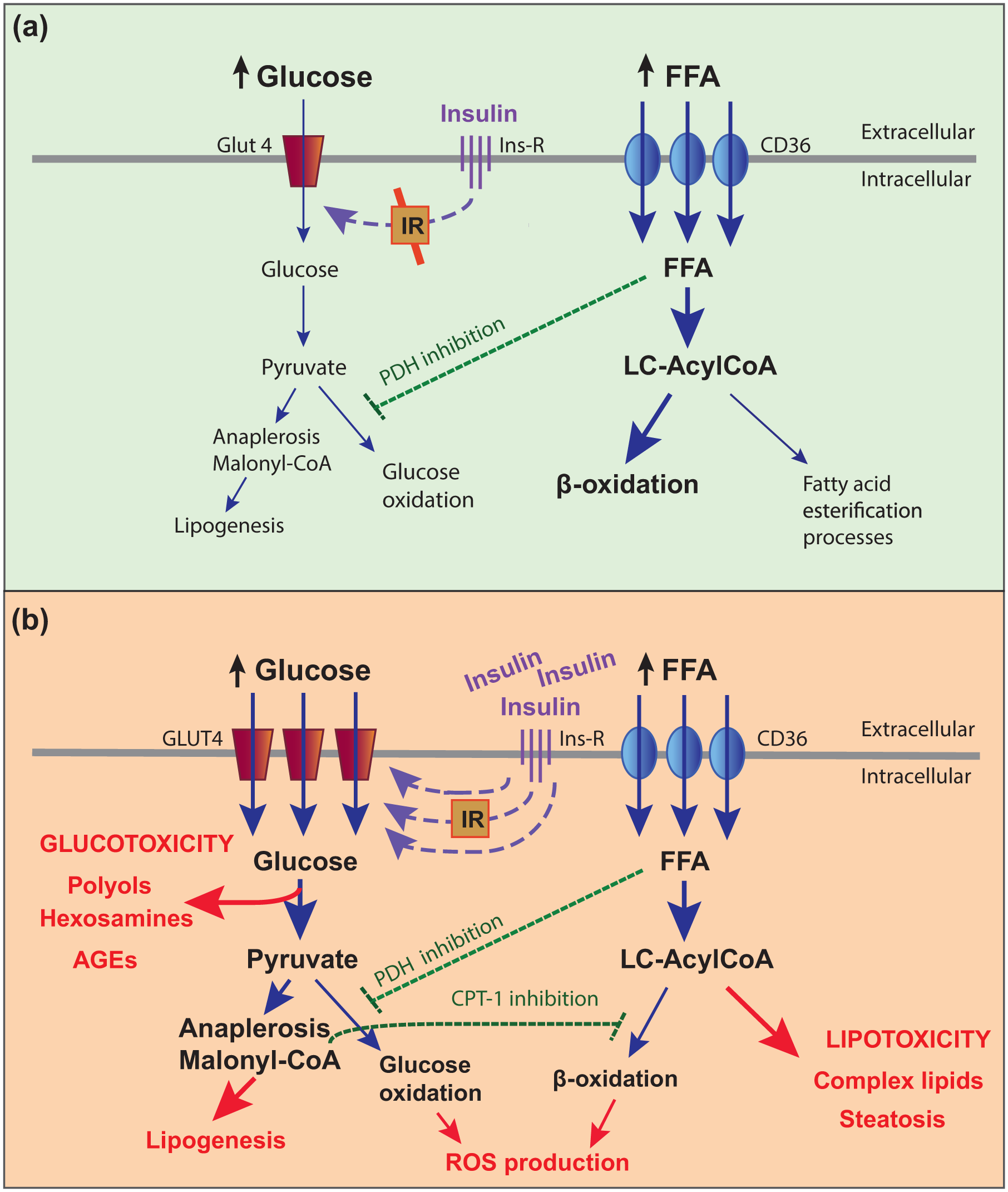
Model illustrating the molecular basis of insulin-induced metabolic stress in obese insulin-resistant and poorly controlled type 2 diabetes patients. Depicted is a cell in which (a) insulin resistance (IR) protects from nutrient overload and metabolic stress by limiting glucose flux into the cell at times when both glucose and free fatty acids (FFA) are elevated in blood; (b) the IR protection is overridden by a high dose of exogenous insulin therapy which promotes excess glucose uptake and both glucotoxicity and lipotoxicity. High FFA availability inhibits glucose oxidation at the level of pyruvate dehydrogenase (PDH), such that a high glucose flux promoted by high levels of insulin will be forced into glucotoxic pathways above this step, including the polyol and hexosamine pathways, as well as the production of advanced glycation end product (AGE) precursors. Furthermore, high glucose availability promotes build-up of cytosolic malonyl-CoA which will inhibit carnitine palmitoyltransferase 1 (CPT1) and the transfer of long-chain acyl-CoAs (LC-AcylCoA) into mitochondria for β-oxidation. This will result in a push of intracellular FFA metabolism towards synthesis of complex lipids, such as diacylglycerols, cholesterol esters and ceramides, and steatosis causing lipotoxicity. Excess glucose supply to the mitochondria in the presence of high FFA supply will also promote reactive oxygen species (ROS) production and oxidative damage.
A failure to limit excess entry of glucose at times of concomitant high free fatty acid (FFA) availability will cause cell injury by the mechanisms of glucolipotoxicity. 12 High FFA availability will inhibit glucose oxidation at the level of pyruvate dehydrogenase, such that a high glucose flux will be forced into pathways above this step, including glycogen synthesis, the polyol and hexosamine pathways, and the production of advanced glycation end product (AGE) precursors (Figure 1).21,22 Similarly, high glucose availability, via malonyl-CoA/AMPK metabolic sensing mechanisms, will inhibit FFA oxidation such that intracellular FFA metabolism will be pushed towards esterification and other processes causing intracellular steatosis and accumulation of complex lipids such as diacylglycerols, cholesterol esters and ceramides (Figure 1).23,24 An excessive mixed nutrient entry into cells will also overload the electron transfer chain resulting in mitochondrial dysfunction and increased reactive oxygen species (ROS) production.25,26 Endoplasmic reticulum stress and activation of the inflammasome are also known consequences of excessive nutrient entry.23,27–29 The concept of ‘insulin-induced metabolic stress’ has been discussed in more detail previously. 15
Islet β-cell role in obesity and T2D: upstream or downstream of insulin resistance?
The predominant view is that islet β-cell failure in obesity-related T2D is a consequence of it not being able to sustain high enough insulin secretion to compensate for insulin resistance, suggesting it is downstream and a victim of insulin resistance. 12 However, increasing evidence from pre-clinical and clinical studies support an alternate possibility, at least in subsets of individuals at risk of T2D, that hyper-responsiveness of the islet β-cell to a hostile environment (e.g. from a westernised lifestyle) drives hyperinsulinaemia, this being the culprit and upstream to excessive weight gain, insulin resistance, subsequent β-cell failure and the development of T2D.30–33
There is considerable heterogeneity in islet β-cell function in mouse strains with those that have a tendency for insulin hypersecretion (e.g. DBA/2 compared to the C57Bl/6 and 129 T2 strains) being more prone to high fat diet–induced weight gain and β-cell failure.33,34 Further-more, there are several examples by which suppression of insulin secretion through genetic manipulation can reduce high fat diet–induced obesity and insulin resistance. Islet β-cell-specific deletion of the adipose triglyceride lipase, through reducing the lipid amplification arm of fuel-induced insulin secretion, protects mice from obesity, hyperinsulinaemia, insulin resistance and hyperglycaemia. 35 In addition, through suppressing insulin secretion by knocking out three of the four insulin gene alleles (Ins1−/−; Ins2+/− and Ins1−/−; Ins2+/+), it has been shown that ageing female mice have lower glycaemia, improved insulin sensitivity and an extended life span. 36 The model less predictably altered insulin secretion in male mice. 30 In the leptin deficient ob/ob mouse model of obesity, a similar genetic approach to lowering insulin secretion, while successfully being able to attenuate obesity, resulted in the development of diabetes, indicative of a need for compensatory hyperinsulinaemia for obesity-related insulin resistance when a rare monogenic cause of obesity rather than hyperinsulinaemia is the primary cause of the excessive weight gain. 37
Of relevance within human studies is the Da Qing Children Cohort Study which showed that fasting insulin at the age of 5 years, after the adjustment for age, sex, birth weight, TV-viewing time and weight (or body mass index) at baseline, predicted weight gain from age 5 to 10 years. 38 Furthermore, higher insulin levels at 5 years of age were also predictive of higher levels of systolic blood pressure, fasting plasma glucose, insulin resistance as determined by the homeostasis model and triglycerides at 10 years of age, all features of the MetS. 38 The findings were similar to those in a study of Pima Indian children. 39 In addition, adolescent girls with PCOS have been shown to have early-onset insulin hypersecretion in association with insulin resistance. 40
Pharmacological approaches to suppress insulin secretion in humans also support the view that hyperinsulinaemia may have more of a primary role in the MetS. In obese men, 6 months treatment of lifestyle change with either diazoxide (DZ) alone (inhibits insulin secretion by activating the ATP sensitive potassium channels), DZ with metformin (DZ + M) or placebo showed that DZ (DZ and DZ + M groups combined) markedly reduced fasting insulin levels by 72% compared to only 23% in the placebo group (p < 0.001), and this was accompanied by greater improvements in body weight, LDL cholesterol, HDL cholesterol, triglyceride, and systolic and diastolic blood pressure. 41 Similar findings were found when hyperinsulinaemia was suppressed by the somatostatin analogue octreotide LAR in obese subjects, with evidence of responders and non-responders to this therapy. 42 Also of relevance, in subjects with T2D, short-term DZ use is capable of restoring islet β-cell function through β-cell rest.43,44
Thus, considerable evidence points to insulin hypersecretion as being at, or close to, the root cause of MetS and its related conditions, with insulin resistance being downsteam. Focus on reducing insulin hypersecretion, at least early in the course of these conditions, is likely to have beneficial metabolic effects.
Towards better stratification of diabetes: subset of severe insulin-resistant and hyperinsulinaemic diabetes
Within a recently reported study of adult-onset diabetes from Scandinavia, five subgroups were identified: severe autoimmune diabetes (SAID), severe insulin-deficient diabetes (SIDD), severe insulin-resistant diabetes (SIRD), mild obesity-related diabetes (MOD) and mild age-related diabetes (MARD). 45
The subgroup that seems most relevant to this discussion is SIRD, with the predominant characteristics being obesity, severe hyperinsulinaemia and insulin resistance. An alternative name for this subgroup could have been ‘severe hyperinsulinaemic diabetes’. Individuals within this subgroup, in keeping with the concept of insulin-induced metabolic stress, were also more likely to develop diabetic nephropathy and have coronary events. 45 Surprisingly, the age of diabetes onset in the SIRD group was relatively high, which may relate to the predominant Scandinavian ethnicity within the diabetes registries used. 45 The SIRD subgroup characteristics of more severe hyperinsulinaemia and insulin resistance tend to be mirrored in young people presenting with obesity-related T2D, as was found in the Restoring Insulin Secretion (RISE) study and is also reported in various high-risk indigenous groups.40,46,47 T2D in youth is also associated with a much higher risk of early-onset nephropathy and macrovascular disease. 40 If in this subset of diabetes (SIRD), insulin hypersecretion rather than insulin resistance has the primary role, as remains to be determined, it will have major implications on the best approaches to prevention and treatment.
In the SIDD, MOD, and MARD subgroups, insulin resistance is of lesser degree at the time of diabetes diagnosis; however, islet β-cell failure must be involved in the pathogenesis. Whether mild suppression of insulin secretion in at least some of those at risk within these subgroups would prevent this β-cell failure and T2D development is unknown. A precision medicine approach will most likely be required in which the correct approach to diabetes prevention and treatment will require detailed phenotypic and genotypic classification of individual patients within these subgroups.
A paradigm shift: new conceptual framework for considering insulin resistance and the MetS
If insulin resistance, while clearly being a biomarker of poor metabolic health, is also to be considered a defensive mechanism used by critical tissues against hyperinsulinaemia and nutrient overload, a complete revision of the conceptual framework within which hyperinsulinaemia, insulin resistance and the MetS are viewed, is needed (Figure 2). Such a revision is not trivial, as it has major implications for how MetS and its associated conditions, including T2D, PCOS, NAFLD and ASCVD, should be prevented and managed. Within this new framework and paradigm shift, hyperinsulinaemia has a more primary or causative role. In doing so, instead of the role of the islet β-cell being one of ‘compensation’ for insulin resistance, it becomes the primary driver, with insulin hypersecretion and the resulting hyperinsulinaemia taking up position as the unifying mechanism. Thus, the development of new therapeutic approaches for MetS, and at least the SIRD subgroup of T2D, will need to move to prevention and/or suppression of the hypersecreting β-cell (Figure 2). Approaches to lower glucose and other elevated nutrients in the blood of MetS and T2D patients through overriding the protective role of insulin resistance will be contraindicated, as we and others have previously advocated.14–17 While research into mechanisms of islet β-cell failure and insulin resistance will continue to be important, more focus on the mechanisms driving insulin hypersecretion will be required, whether they be genetic or acquired, including those acquired early in life from epigenetic processes and/or consequent on islet β-cell response to new environmental exposures. 31
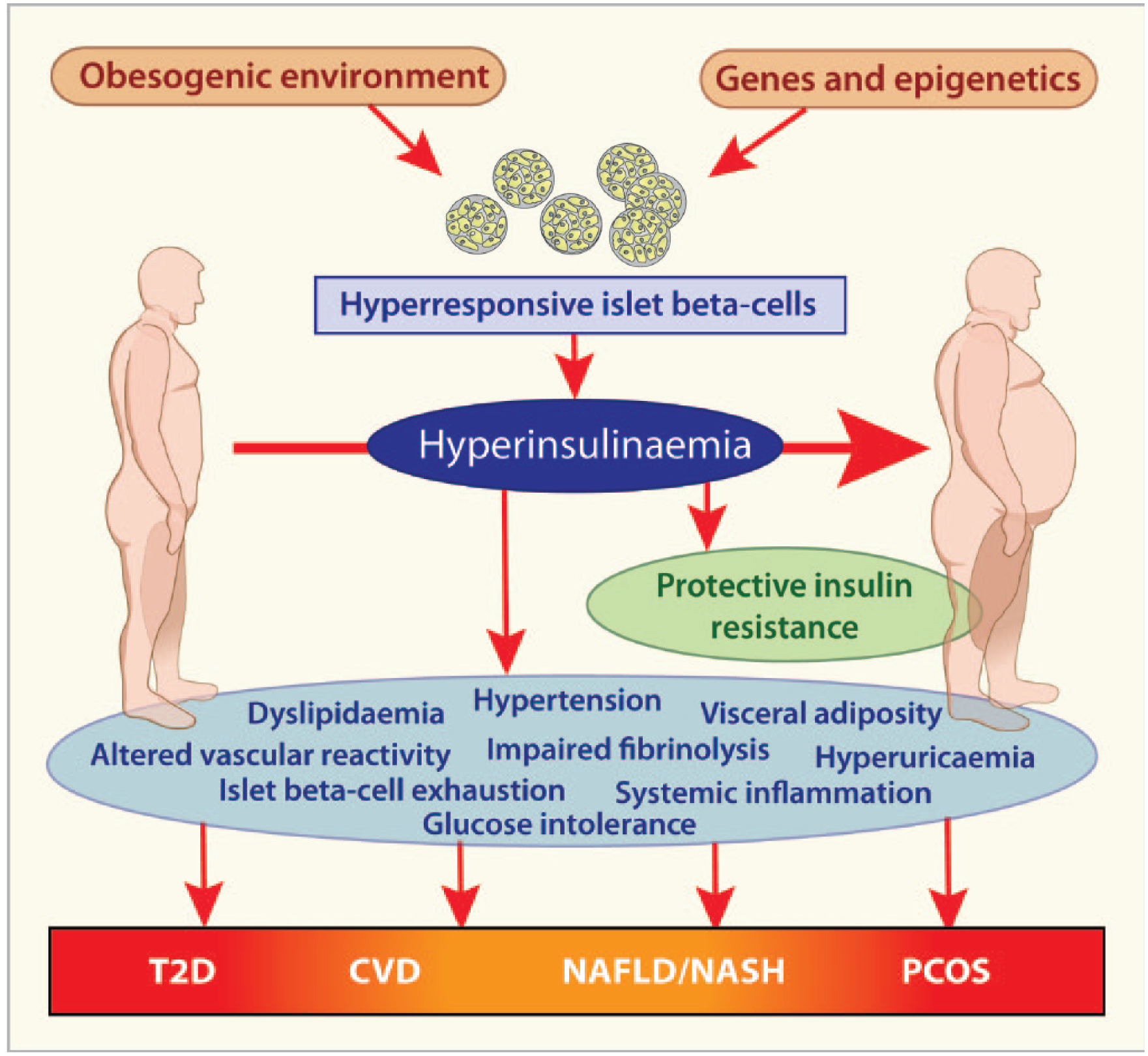
A new conceptual framework for considering insulin resistance and the metabolic syndrome (MetS) and its associated disorders. The key changes in this conceptual framework are the roles of hyperinsulinaemia and insulin resistance in the MetS. Islet β-cell hyper-responsiveness to adverse environmental factors in genetically or epigenetically predisposed individuals results in hyperinsulinaemia and this is the primary driver of the MetS. Insulin resistance provides protection for critical tissues against insulin-induced metabolic stress and, while being a biomarker of metabolic ill health, is not a driver of pathogenesis. The characteristic features of MetS are driven by hyperinsulinaemia. The MetS-related disease entities, type 2 diabetes (T2D), cardiovascular diseases (CVD), non-alcoholic fatty liver disease/non-alcoholic steatohepatitis (NAFLD/NASH) and polycystic ovary syndrome (PCOS) are a downstream consequence of hyperinsulinaemia and the MetS.
Relevance to management of T2D
Optimisation of cellular nutrient status
In managing disturbed metabolic homeostasis in T2D, the focus of clinicians is currently on normalising glucose and lipid parameters in the blood. Less thought is given to optimising intracellular metabolism, even though nutrient-induced tissue injury in obesity-related T2D is predominantly a consequence of excess entry of nutrients from the blood into cells. This is understandable, as measuring nutrient levels is much easier in blood (e.g. blood glucose, HbA1c and plasma triglycerides) than in cells. The corollary is that approaches to normalise glycaemia in obesity-related T2D that drive glucose and other nutrients into already nutrient-overloaded cells, such as by high-dose insulin therapy or sulphonylureas to override insulin resistance, or insulin sensitisers to reverse insulin resistance depending on mechanism of action, may unintentionally cause harm. 15 According to this argument, alternative approaches to lowering glycaemia that nutrient off-load cells, such as intensive lifestyle measures, sodium-glucose transporter 2 (SGLT2) inhibitors, glucagon-like peptide-1 receptor agonists or bariatric surgery, should be beneficial in the majority of patients with obesity-associated T2D (Figure 3(a)). 15
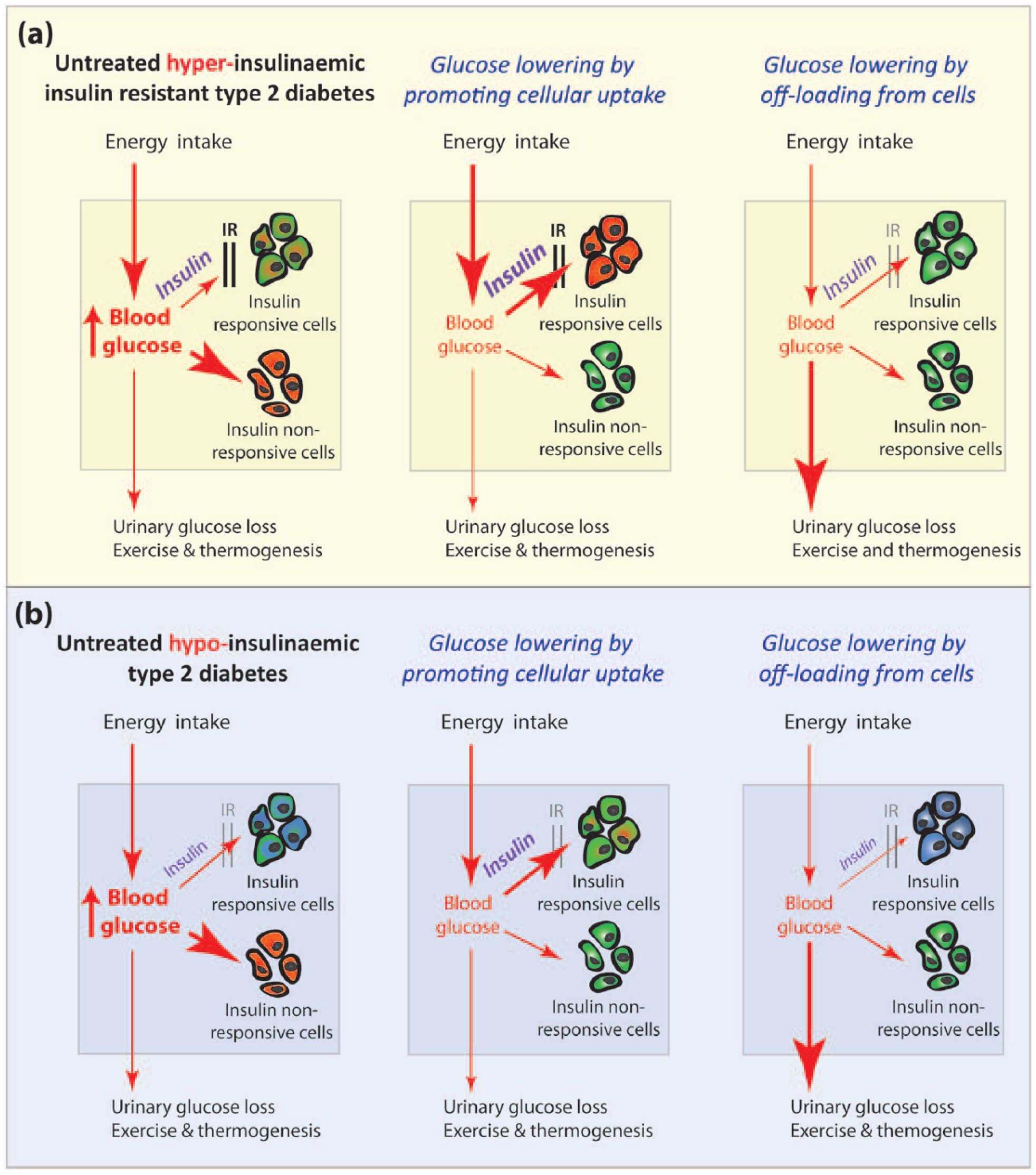
Optimisation of cellular nutrient status in patients with hyper- or hypoinsulinaemic type 2 diabetes: importance of the approach to glucose lowering. (a) In untreated type 2 diabetes (T2D) with hyperinsulinaemic diabetes, insulin resistance (IR) protects insulin-responsive cells such as cardiomyocytes and skeletal muscle cells from nutrient overload; cells such as endothelial cells that are non-responsive to insulin with respect to glucose uptake, however, are not protected and are injured by glucotoxicity contributing to diabetes complications (left panel). Glucose-lowering approaches that override the physiological IR to force glucose into insulin-responsive tissues (e.g. by high-dose insulin therapy) may reduce glucotoxicty in some tissues, but at the cost of nutrient-induced injury to the insulin-responsive tissues (e.g. causing a metabolic cardiomyopathy) (centre panel). Glucose-lowering approaches that off-load glucose from cells of critical body tissues, by either reducing glucose entry into the blood (e.g. intensive lifestyle, α-glucosidase inhibitors and bariatric surgery) or glucose clearance from the blood via non-toxic routes (e.g. SGLT2 inhibitors to promote urinary glucose loss, exercise and activation of brown adipose tissue), will reduce nutrient-induced tissue injury in all cell types (right panel). (b) In untreated hypoinsulinaemic T2D, a starvation response to insulin deprivation occurs in which free fatty acid (FFA) release from adipose tissue is increased and hepatic ketone body production is increased; cardiomyocytes and skeletal muscle cells are starved of glucose and switch to use FFA and ketone body for energy; cells such as endothelial cells are not protected from hyperglycaemia (left panel). In this circumstance, insulin therapy will prevent the starvation response and normalise cellular nutrient status in insulin-responsive tissues and prevent glucotoxicity in endothelial cells (centre panel). Glucose-lowering approaches that off-load glucose from cells (e.g. by very low carbohydrate diets or fasting for surgery) together with glucose clearance from the blood (e.g. SGLT2 inhibitors) will exacerbate the starvation response in hypoinsulinaemic T2D and induce euglycaemic ketoacidosis (right panel). Healthy cells are shown in green; unhealthy nutrient-overloaded cells are shown in red; and unhealthy nutrient-deprived cells are shown in blue.
The alternate scenario of intracellular nutrient depletion in patients with hypoinsulinaemic diabetes is also important to consider, particularly, with the increasing occurrence of cases of euglycaemic ketoacidosis in patients treated with SGLT2 inhibitors. 48 Avoidance of SGLT2 inhibitors and most often a shift to insulin therapy will be necessary in such patients (Figure 3(b)).
Thus, the approach to diabetes management should take into account some consideration of cellular nutrient status (Figure 3). For these reasons, new blood biomarkers of cellular nutrient or energy status may also be of value in patient care and once discovered should be examined for clinical utility.
In support of the proposition that glucose-lowering approaches that work by driving glucose into tissues can be harmful in overweight and obese subjects with T2D and insulin resistance, we reviewed major T2D clinical trials and found that whenever intensive glucose-lowering approaches were associated with weight gain of greater than 1.0 kg/year [Action to Control Cadiovascular Risk in Diabetes (ACCORD), Veterans Affairs Diabetes Trial (VADT) and Diabetes Mellitus Insulin-Glucose Infusion in Acute Myocardial Infarction 2 (DIGAMI 2)], cardiovascular and all-cause mortality increased, although only reaching statistical significance in ACCORD given the greater sample size. 15 Furthermore, among adults with diabetes and stable ischaemic heart disease aged ⩾75 years, insulin provision therapy was associated with an increased risk for all-cause mortality [hazard ratio = 1.89, confidence interval (CI) = 1.1–3.2, p = 0.020]. 49
In support of the benefits of nutrient off-loading approaches are more recent clinical trials of new classes of glucose-lowering agents, such as SGLT2 inhibitors (by promoting urinary glucose loss) and GLP-1 receptor agonists (by reducing weight through increased satiety), as well as bariatric surgery that have demonstrated reductions in major adverse cardiovascular and renal outcomes in high-risk T2D patients.50–52 The recent consensus statement of the American Diabetes Association (ADA) and European Association for the Study of Diabetes (EASD) on the management of hyperglycaemia in T2D has taken the results of these major clinical trials into consideration in their recommendations. 53
Prevention of insulin hypersecretion
The nutrient off-loading approaches to glucose lowering available in the management of T2D, including intensive lifestyle change, SGLT2 inhibitors, GLP-1 receptor antagonists, α-glucosidase inhibitors and bariatric surgery, will all reduce insulin hypersecretion. However, often these therapies are started once T2D is established and failure of islet β-cells has already commenced. Optimal approaches for reversal of severe hyperinsulinaemia in patients prior to development of T2D or early in its course, in particular in younger individuals, when lifestyle measures are generally unsuccessful, are not known. Of note, bariatric surgery has been shown to be effective in reversing hyperinsulinaemia and MetS in obese adolescents. 54 In the RISE study, neither 3 months of insulin glargine followed by 9 months of metformin nor 12 months of metformin alone slowed the progressive deterioration of β-cell function in young people with early T2D, suggesting different approaches are required. 55 The development of specific islet β-cell therapies to limit insulin hypersecretion in high-risk individuals with MetS-related conditions and obesity-related pre-diabetes and early T2D should be pursued.
Conclusion
The part played by Jerry Reaven in linking the dots between the various components of the MetS and the relevance of MetS to ASCVD, NAFLD, PCOS and T2D has been enormously important. The search for the unifying mechanism has been contentious. Here, we make a case for putting ‘insulin hypersecretion’ into this role, while considering insulin resistance as a protective downstream response. This necessitates a complete revision of the conceptual framework within which we view insulin resistance and the pathophysiology of the MetS and obesity-associated T2D, which if confirmed, has major implications for the prevention and management of these metabolic conditions.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: C.J.N. has received speaking fees from AstraZeneca, Servier and Sanofi; M.P. declares no conflicts of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported in part by grants from the Canadian Institutes of Health Research (M.P.) and the National Health and Medical Research Council [project grant 1128442 (C.J.N.)].