
Abstract
Sarco(endo)plasmic reticulum calcium adenosine triphosphatase is responsible for transporting cytosolic calcium into the sarcoplasmic reticulum and endoplasmic reticulum to maintain calcium homeostasis. Sarco(endo)plasmic reticulum calcium adenosine triphosphatase is the dominant isoform expressed in cardiac tissue, which is regulated by endogenous protein inhibitors, post-translational modifications, hormones as well as microRNAs. Dysfunction of sarco(endo)plasmic reticulum calcium adenosine triphosphatase is associated with heart failure, which makes sarco(endo)plasmic reticulum calcium adenosine triphosphatase a promising target for heart failure therapy. This review summarizes current approaches to ameliorate sarco(endo)plasmic reticulum calcium adenosine triphosphatase function and focuses on phospholamban, an endogenous inhibitor of sarco(endo)plasmic reticulum calcium adenosine triphosphatase, pharmacological tools and gene therapies.
Keywords
Introduction
Calcium (Ca2+) is the most pervasive carrier of signals and involved in many fundamental cell processes, such as muscle contraction.1,2 Ca2+ homeostasis in cardiomyocytes is maintained mainly by Ca2+ ion channels, especially Ca2+ adenosine triphosphatase (ATPase). Three types of Ca2+ pumps have been discerned in higher animals: sarco(endo)plasmic reticulum calcium adenosine triphosphatases (SERCAs), 3 plasma membrane calcium adenosine triphosphatases (PMCAs) 4 and secretory-pathway calcium adenosine triphosphatases (SPCAs). 5 All of these Ca2+ pumps belong to the family of P-type ATPases, which are characterized to couple hydrolysis of adenosine triphosphate (ATP) to the movement of ions across biological membrane. 6
SERCA is the main Ca2+ pump for the cytoplasmic Ca2+ homeostasis in cardiac muscle, which is responsible for the reuptake of Ca2+ from the cytosol back into sarcoplasmic reticulum (SR) lumen during the relaxation of the cardiac muscle. 7 Distinguishable isoforms of SERCA have been identified in different tissues, while SERCA2a is abundantly expressed in cardiac muscle and mainly accountable for maintaining intracellular Ca2+ cycling as well as regulating cardiac contraction. It has been proved that reduced rate of SR Ca2+ uptake, lower expression and function of SERCA2a are associated with various cardiac diseases, including ischaemic heart disease, hypertrophic and dilated cardiomyopathy that often progress to heart failure.8–11 In this review, we aim to discuss the physiological function of SERCA2a and its role in heart failure and focus on the promising therapeutic approaches to heart failure via targeting SERCA2a.
Isoforms of the SERCA pump
SERCA has been detected in various living organisms, from yeast to mammalian systems. In vertebrates, three main SERCA proteins are encoded by three different genes (ATP2a1/SERCA1, ATP2a2/SERCA2, and ATP2a3/SERCA3).12,13 More than ten variants are produced by tissue-dependent alternative splicing at the COOH-terminal of the transcripts,2,14,15 which result in specific functions of the variants.
SERCA1, expressed in fast-twitch skeletal muscle, is found possessing two major splice variants: SERCA1a (adult form) and 1b (neonatal form).16,17 In addition, another gene splice pattern (S1T), a cis-activate SERCA1 transcripts, is discovered in liver and several other human adult and foetal tissues, but S1T is unavailable in muscle. 18
Two disparate tissue-specific SERCA2 isoforms (SERCA2a and 2b) are derived from the alternative processing of SERCA2 gene transcripts in COOH-terminal. 19 SERCA2a, also known as ‘cardiac isoform’, is the dominant isoform in cardiac muscle and slow-twitch skeletal muscle,20,21 while it is also found at lower amounts in smooth muscle and neuronal cells. 22 SERCA2b is traceable in almost all tissues including muscle and nonmuscle cells as a housekeeping isoform. Recently, a third splice variant (SERCA2c) is reported expressed in the left ventricle, which may be functionally important in (foetal) heart.23,24 The above mentioned variants possess identical sequence of the first 993 amino acids, while 49-residue-long peptide chain in SERCA2b and six amino acids in SERCA2c occupied four amino acids (tetrapeptide tail) in the COOH-terminal of SERCA2a protein. The different C-terminal tail of the three isoforms accounts for their functional differences. Expression of SERCA isoforms is not only tissue specific but also undergoes developmental regulation, with switching among the isoforms involved. In the heart, SERCA2a expression exceeds during development and in adult stages.25,26 Lately, a new splicing variant (SERCA2d) is discovered in human skeletal muscles, 19 yet more endeavour is needed to explore the corresponding physiological meaning.
SERCA3a is known as the major SERCA3 isoform, 27 which has additional five variants (SERCA3b-f) in humans,27–30 two in mice (SERCA3b and c) 31 and one in rats (SERCA3b). 32 SERCA3 is widely expressed in many tissues at various concentrations, which is prominent in haematopoietic cell lineages, platelets, epithelial cells, fibroblasts, as well as endothelial cells, but only a minority of SERCA3 is expressed in muscle,14,27,33,34 and it is found invariably to colocalize with SERCA2b in each of the cell types. 35
A notable feature addressed here is highly conserved primary structure of these SERCA patterns, which resulted in identical transmembrane (TM) topologies and tertiary structures. Toyoshima et al. 36 described the crystal structure of SERCA1a at 2.6 Å resolution and identified the existence of cytoplasmic region (consisted of an actuator or anchor domain, phosphorylation and adenosine-binding site) and TM domain that are organized with α-helices (M1–M10).37,38 Using oligo-nucleotide–directed site-specific mutagenesis, Clarke et al. 39 identified that Ca2+-binding sites of SERCA are close to the centre of the transmembrane domain (M4, M5, M6, and M8), and it is accessible only from the cytoplasm. 2 SERCA is located on endoplasmic reticulum (ER) membrane and is responsible for the Ca2+ uptake into ER in order to maintain ER Ca2+ homeostasis. Generally, SERCA has two major conformational states: E1 (high Ca2+ affinity states) and E2 (low Ca2+ affinity states). 40 As predicted, SERCA is capable to transfer the terminal phosphate of ATP to an aspartate residue to transport Ca2+across the membrane (Figure 1). Before translocation, cytosolic portion of the pump switches to an ‘open’ state and two Ca2+ bound to the M domain side by side with high affinity (E1· 2Ca2+), which triggers the hydrolysis of ATP and phosphorylation of the pump. The enzyme then undergoes a series of conformational changes, during which the cytosolic compartment gains a ‘compact’ state with low affinity to Ca2+ (E2) and faces the luminal side of the membrane. Ca2+ then dissociates from the complex (E2· 2Ca2+), followed by the dephosphorylation of the pump. E2 finally goes through conformational changes to E1 and then it is recruited for the Ca2+ transfer.
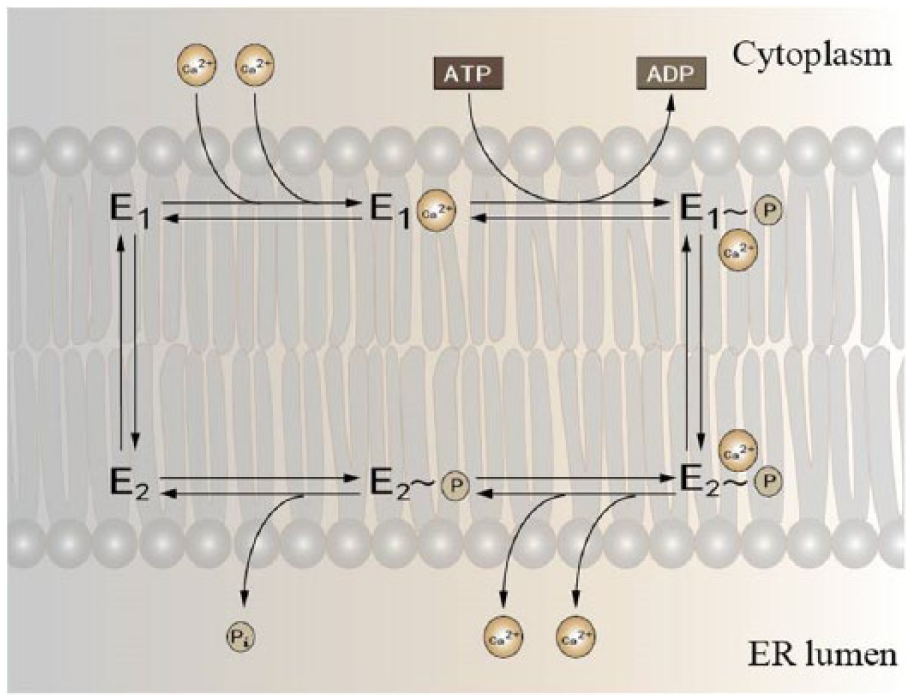
Illustrating of Ca2+ transport recycling in cardiac muscle. SERCA pump possesses two main conformational states: E1 and E2. E1 has a high Ca2+-binding affinity at the cytosolic site, while E2 binds to Ca2+ with low affinity and thus delivers it to the opposite site of the membrane against the concentration gradient with the aid of hydrolyzing one ATP molecule for each two Ca2+.
Physiological activity of SERCA2a and regulators in the heart
SERCA regulates intramuscular Ca2+ handling during contraction relaxation in cardiomyocytes
As a ubiquitous second messenger, Ca2+ flow is essential in excitation–contraction (EC) coupling of cardiac muscle and directly activates the myofilaments, which causes muscle contraction. During EC coupling, depolarization of the membrane triggers voltage-gated Ca2+ channels open at relatively small amount of Ca2+ influx. 41 As illustrated in Figure 2, in normal cardiomyocytes, the voltage-dependent L-type calcium channel (LTCC), physically accumulates at the transverse tubules (t-tubules), serves as the primary source of Ca2+ and allows entry of Ca2+ into the myocyte. The increased concentration of intracellular Ca2+ further initiates the opening of ryanodine receptor 2 (RyR2) which triggers the local release of Ca2+ from SR – the major Ca2+-storing organelle in cardiomyocyte – resulting in the contraction of cardiac muscle. 42 During the process of Ca2+-induced Ca2+ release, cytosolic Ca2+ concentration raises from 0.1–0.2 μM to 2.0–10.0 μM. 43 Then cytosolic Ca2+ is pumped back to SR by SERCA or extruded by sarcolemmal sodium ion (Na+)/Ca2+ exchanger (NCX) and induces relaxation of muscle,7,41 while PMCA exerts only minor function in lowering the Ca2+ level in cardiomyocytes. In higher mammalian species and human, nearly 75% of the cytosolic Ca2+ is eliminated by SERCA and 25% by NCX, while their contributions vary among species. ATP hydrolysis is required for the translocation of two Ca2+ into SR lumen via SERCA against the approximately 104-fold concentration gradient. 44
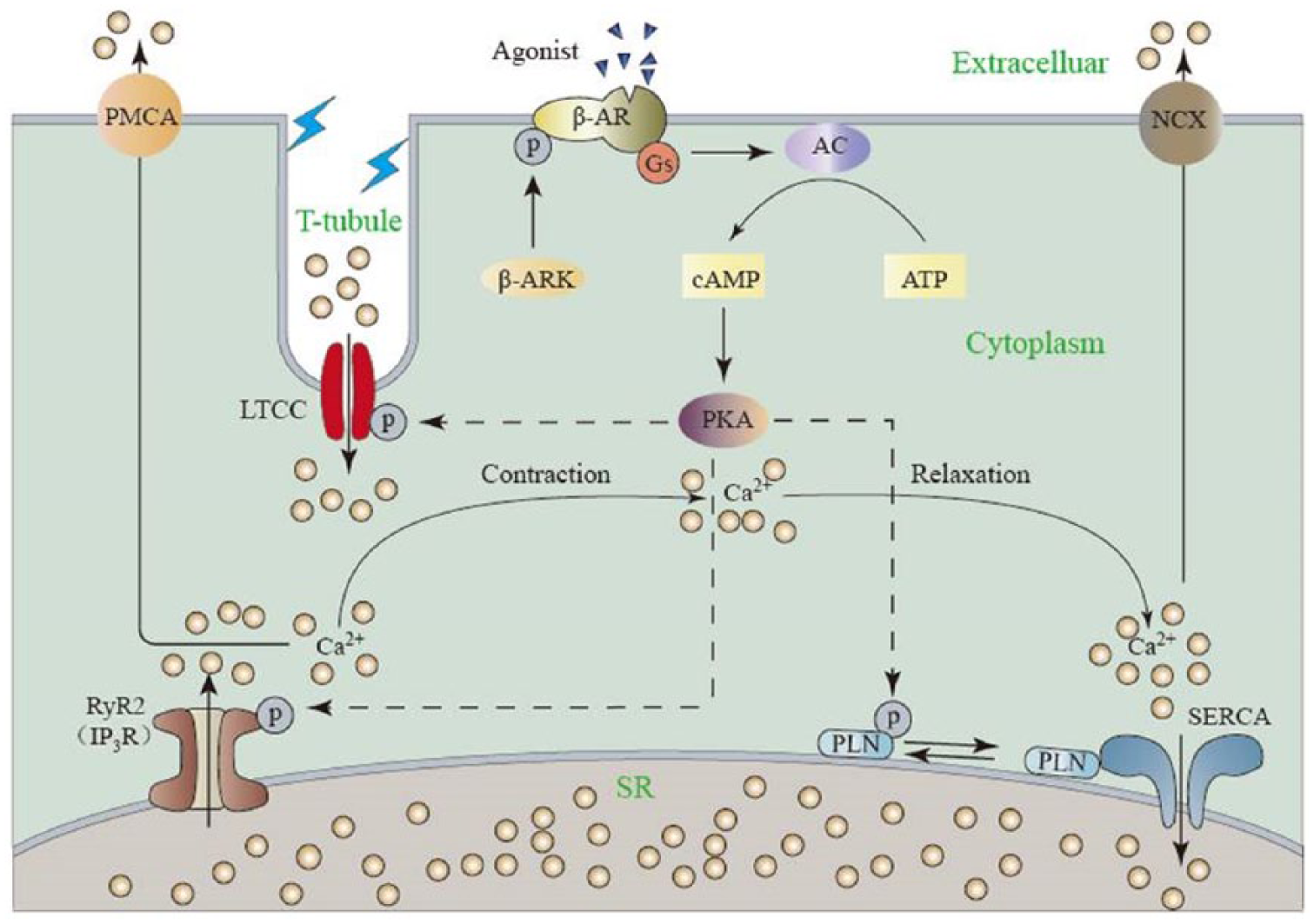
Schematic of Ca2+ handling in cardiomyocyte during excitation–contraction coupling. A small amount of Ca2+ uptake via LTCC lead to the release of a large amount of Ca2+ from SR via RyR2, resulting in the contraction of myofilaments. Relaxation occurs when sequestration of Ca2+ back into SR by SERCA or NCX. LTCC: L-type calcium channel; RyR2: ryanodine receptor 2; IP3R: inositol 1,4,5-trisphosphate (IP3) receptor; NCX: Na+/Ca2+ exchange; PLN: phospholamban; SR: sarcoplasmic reticulum.
Cardiac contractility is controlled by the amplitude and kinetics of Ca2+ handling in cardiomyocytes, which in turn are regulated by phosphorylation and dephosphorylation of key proteins involved in EC coupling (Figure 2). G protein–coupled β-adrenergic receptor (β-AR) is able to activate adenylyl cyclase, which further catalyzes cyclic adenosine monophosphate (cAMP) production and protein kinase A (PKA) activation. PKA is the main source of phosphorylation of various proteins involved in EC coupling in cardiac muscle, including LTCC, RyR2, inositol 1,4,5-trisphosphate receptor (IP3R) and phospholamban (PLN). Simultaneously, a small amount of the key targets are also phosphorylated by Ca-calmodulin-dependent protein kinase (CaMKII) at different molecular sites. 45 Phosphorylation of the above mentioned proteins (e.g. RyR2) may induce increased amplitude and velocity of Ca2+ removal and reuptake and even enhanced contractility. 46 PKA is therefore considered as the ‘on switch’ for contractility, while some related phosphatases that hydrolyzes cAMP serves as ‘off switch’.
In SR, Ca2+ bind to Ca2+-binding protein calsequestrin predominantly and are stored close to the release channel (e.g. RyR2), 47 which may facilitate the Ca2+ availability during early systole. SERCA2a is the main player to restore diastolic Ca2+ and terminate Ca2+-dependent activation, while some other components, such as phosphorylation of RyR2 or IP3R, can regulate the SR Ca2+ release mechanism. Several direct or indirect factors have been found to have crucial modulation effect on the function of SERCA2a. Among them, PLN is accepted as the most important prevailing indirect regulator of cardiac isoform SERCA2a/b,48,49 dephosphorylation form of which presents apparent inhibition to Ca2+ affinity of SERCA2a, while there are also some other components that can directly regulate SERCA expression and activity, which will be discussed in detail.
Regulation of SERCA2a expression and activity in the heart
SERCA2 is expressed abundantly in adult mammalian heart. SERCA2a is validated to be the major cardiac isoform (~99.9%), while ubiquitous SERCA2b represents less than 0.1% of total SERCA mRNA. 50 Moreover, the relative level of SERCA2a transcripts lessens severely in both animal models of heart failure and cardiac patients, while no significant change is observed in the level of SERCA2b during heart development. Owing to its pivotal role in regulating cardiac contractility, in this section we summarize several important regulators of SERCA2a, including the endogenous protein inhibitors, post-translational modifications, hormones as well as the microRNAs (miRNA).
The role of PLN and sarcolipin in regulating SERCA2a
PLN and sarcolipin (SLN) are small homologous SR or ER membrane intrinsic proteins, consisting of 52 and 31 amino acid residues respectively, both of which can inhibit activity of SERCA by lowering the pump’s apparent affinity for Ca2+. PLN is predominantly expressed in cardiac muscle, while SLN is abundant in skeletal muscle. The tertiary structure of monomeric PLN is described as ‘L-shaped’ consisting of N-terminal cytosolic domain IA, flexible linker, domain IB and C-terminal TM domain II, which is essential for PLN oligomerization.51–53 When the amount of Ca2+ in systole reaches a higher level, PLN may oligomerize to form homopentamers owing to the changes of protein’s isoelectric point (from 10 to 6.7),54,55 and three distinct sites of PLN can be phosphorylated: (1) serine-16 (Ser 16 ) residue by cAMP-dependent PKA; 56 (2) threonine-17 (Thr 17 ) residue by CaMKII; 48 (3) serine-10 residue by Ca2+-activated phospholipid-dependent protein kinase (PKC).57,58 Among them, cAMP-dependent phosphorylation site (Ser16) is the most important mediator of PLN activity in transgenic mouse model. 56 Phosphorylation of PLN by the protein kinase increases affinity of SERCA for Ca2+ and relaxation velocity in cardiac muscle and thereby alleviates inhibitory function of PLN. 53
Likewise, SLN is comprised with a TM domain that contains two Ca2+-binding sites, a shorter cytoplasmic region than PLN and luminal domains. 53 Significant sequence homology between SLN and PLN has been discovered in their TM region, suggesting a similar manner of binding to SERCA and alteration of the apparent Ca2+ affinity. 53 SLN phosphorylation at threonine-5 (Thr 5 ) by serine/threonine kinase 16 (STK16) induces the dissociation of SLN from SERCA2a, 59 which has been further confirmed by the results that the inhibitory effect of SLN is relieved during β-adrenergic stimulation in a PLN-null but SLN-overexpressed transgenic mouse model. 60 Moreover, co-expression of SLN and PLN is found in the atria of the heart, in which PLN-SLN complexes lead to super-inhibition of SERCA in smaller rodents, but larger animals are excluded. 61
Role of post-translational modifications to regulate SERCA2a
Several post-translational modifications, including glutathiolation, nitration, SUMOylation, acetylation, glycosylation, as well as O-GlcNAcylation, are also reported directly to regulate the activity of SERCA2a.
During glutathiolation, a disulphide bond is formed between the glutathione and cysteine-674 (C674) residues in SERCA, leading to the enhanced activity of SERCA and thereby increased Ca2+influx.62–64 In general, glutathiolation of SERCA is reversible; however, during oxidant stress such as hyperglycemia in diabetes and atherosclerotic conditions, C674 is oxidized irreversibly, which prevents the glutathiolation of SERCA and inhibits the redox-dependent SERCA stimulation.62,64,65
Nitration is another type of post-translational oxidative modifications. A nitro group was chemically bonded to a protein through nitration due to the exposure to peroxynitrite. 66 A polyol pathway has been put on the map as the main contributor for SERCA2a nitration under hyperglycemic conditions. 67 In failing human hearts, increased inducible NO synthase (iNOS) as well as the end-products of oxidative stress has been detected, 68 and animal models with heart failure exhibit elevated levels of superoxide. 69 All of these findings can raise the nitrotyrosine levels of SERCA nitration, thereby leading to the impaired function of SERCA in human heart failure. 70 However, the regulatory role that how nitration regulates SERCA2a in human is not well elucidated.
Small ubiquitin-like modifier type 1 (SUMO1) can be conjugated to the residues lys480 and lys585 of SERCA2a, which is essential for preserving SERCA2a activity in mouse and human cells. 71 Kho et al. 71 point out that the levels of SUMO1 and SERCA2a SUMOylation decreased significantly in a murine model of pressure overload-induced heart failure. In this work, the restitution of SUMO1 by gene delivery restore SERCA2a protein content and improved cardiac function in heart failure mice, indicating the apparent cardioprotective property of SUMOylation. Moreover, small-molecule activator targeting SERCA2a SUMOylation can promote the contractile properties of cultured rat cardiomyocytes and advance the ventricular function significantly in mice with heart failure. 72 This result is further confirmed by the SUMO1 gene transfer in a swine model of ischaemic heart failure. 73 Even though, more work is urgently needed to uncover the precise role of SUMOylation in rescuing SERCA2a function.
Acetylation of SERCA2a, which may occur at lys464, lys510, and lys533 residues in the nucleotide binding domain of SERCA2a,38,74 can influence intracellular Ca2+ handling in the heart. SERCA2a is found to be acetylated prominently in failing hearts, which is reversible in the presence of silent information regulator 1 (SIRT1) deacetylase. 71
Glycosylation is able to reduce the activity of SERCA2a and Ca2+ efflux during myocardial contraction. SERCA2a is susceptible to be glycosylated exposed to elevated glucose levels,75,76 which has attracted much interest recently in study of the crosstalk between heart failure and diabetes. Glycosylation of SERCA2a increased significantly during diabetes, accompanied with decreased mRNA and protein levels of SERCA2a.75–77
Protein O-GlcNAcylation is mediated by O-GlcNAc transferase,75,78 during which O-linked N-acetylglucosamine is either added or removed from a Ser or Thr residue, and increased O-GlcNAcylation is always associated with aging, 79 ischemia, 80 heart failure81,82 as well as diabetes.75,83 O-GlcNAcylation regulates SERCA2a either by directly influencing SERCA2a expression or altering the phosphorylation of PLN, 83 accompanied with the impaired Ca2+ sequestering.75,84
Role of hormone in SERCA2a regulation
Thyroid hormones (T3) have been well documented considering as important modulator of SERCA1 and SERCA2 at translational level.85,86 A series of studies have illustrated that SERCA2a mRNA expression increases 80%–167% by administering T3 to rodent and rabbit cardiomyocytes, leading to elevated SR Ca2+ uptake and accelerated relaxation time.85–87 Besides the direct modulation of SERCA2a mRNA, T3 also influences the activity of SERCA2a by affecting the mRNA expression of PLN. In various animal models, 61%–75% decreased PLN mRNA has been observed under conditions of hyperthyroidism, and it mounts up to 135% under conditions of hypothyroidism.87–89
Adiponectin is an adipocyte-derived peptide hormone that can regulate the activity of SERCA2a. 90 Adiponectin shows efficient cardioprotective effect by facilitating recovery of SERCA2a activity through lowering inflammation and ameliorating ER stress in animal models with heart failure.90,91 The regulation mechanism of adiponectinon is further investigated, during which increased phosphorylation level of PLN is always viewed, 90 although the precise role remains mysterious. Furthermore, adiponectin can also affect on the signalling of adenosine monophosphate–activated protein kinase (AMPK), 92 which plays a pivotal role in energy homeostasis and induces the alteration of SERCA1a protein and SERCA2a mRNA. 93
Role of miRNA in regulating SERCA2a function
MiRNA is regarded as one of the SERCA2a regulators, recognition site of which is located in the 3′ untranslated region of SERCA2a. Many miRNAs demonstrate distinct effects on the activity of SERCA2a. For example, cardiac ablation of miR-22 in mice can reduce cardiac expression level of SERCA2a and prolonged Ca2+ cytosolic decay. 94 Global analysis reveals miR-22 is likely to affect the expression of many mRNA targets and carries out as a regulator of SERCA2a and myofibrillar protein during stress in the heart. On the contrary, overexpression of endogenous miR-25 results in decreased SERCA2a and contributes to a dramatical loss of contractile function. 95
Modified expression and activity of SERCA2a in heart failure
Although the expression level of SERCA has been observed heterogeneously in failing hearts, reduction of the SERCA2a protein level (and/or activity) with consequential decrease in SR Ca2+ uptake is generally accepted to be of indispensable in the development of failure condition, such as abnormal SR Ca2+ sequestering, inefficient energy expending, as well as impairment of systole and diastole.96–98 Depleted SERCA2a expression or activity inevitably facilitates the deterioration of cardiac function after injury, although it may not be the initial cause of heart disease. 99 To further investigate the physiological relevance of SERCA in cardiac function, many transgenic animal models with SERCA2a overexpression or deficient in the heart have been constructed. Transgenic mice overexpressing SERCA2a exhibit enhanced Ca2+ transients and accelerated contraction–relaxation cycles.100–103 Intriguingly, only modest increase in SERCA2a protein level is obtained despite high levels of mRNA in the genetically modified mice. In addition, transgenic rat lines are established, in which SERCA2a cDNA, expressed specifically in the myocardium, gives rise to significantly increased SERCA2a protein levels,104,105 and the SERCA2a-overexpressed rats demonstrate enhanced intracellular Ca2+ stores, while its contribution in heart failure patients remains to be determined. To gain better understanding of the physiological function of SERCA2a, a mouse model carrying a null mutation in the SERCA2 gene is developed. As expected, homozygous mutants are not obtained, while the heterozygous mice with one single functional allele are reproduced well, showing reduced SERCA2 level, decreased SR Ca2+uptake and significantly lower cardiomyocyte contractility. 106 Although no cardiac hypertrophy or other signs of heart failure is observed in heterozygous mutants, they are hyper-sensitive to pressure overload in an accelerated pathway to heart failure.106–108 In this case, reduced SERCA2a expression has been deemed as the hallmark of heart failure; however, there is also controversial evidence that the protein levels of SERCA2 in failing hearts are similar to that in non-failing hearts of human 109 and animals,110,111 while its activity and rate of Ca2+ uptake are significantly decreased. Simultaneously, SERCA2a contents or activity and impaired SR Ca2+ uptake have been observed in patients with major cardiac diseases such as ischaemic cardiac muscle, hypertrophic heart and dilated cardiomyopathy,112–114 which often then progress to heart failure. Following this line, therapeutic approach targeting SERCA2a is potential to alleviate the deficient contractile function in heart failure patients.
SERCA2a as an efficient therapeutic target for heart failure
Accumulating evidence proves that the expression and function of SERCA2a decrease in many cardiac diseases including myocardial hypertropy and dilated cardiomyopathy in both of experimental animal models and human patients. Over the past decades, several therapeutic approaches have been widely authenticated to prevent the reduction and calibrate SERCA2a-mediated Ca2+ uptake in heart failure, 97 and SERCA2a has been one of the most promising targets for recovery of cardiac contraction.
Reducing SERCA2a inhibition by targeting PLN (indirectly)
PLN has been generally recognized as the critical factor for regulating Ca2+ reuptake, since the interaction between PLN and SERCA may inhibit the pump’s activity. 53 Diverse level of PLN are detected in the heart, which may contribute to different contractile property through influence on SERCA2a. 115 In failing hearts of human and experimental animals, the amount of PLN protein decreases dramatically;116–118 while in some other studies expression level of PLN protein is unchanged, yet PLN phosphorylation is much lower than that in non-failing hearts, which accounts for the further inhibition of SERCA2a activity. 97 Given that knockout of PLN is able to alleviate the impairment of cardiac contraction and morphological change with dilated cardiomyopathy in rodent model, 115 attenuating PLN-mediated inhibition of SERCA2a is a promising strategy for the treatment of heart failure. In mice cardiomyocytes, enhanced SERCA activity and increased SR Ca2+ load are obtained with a muscle-specific long noncoding RNA by displacing the SERCA inhibitors, PLN, SLN and myoregulin (MLN), which represents an attractive means of enhancing cardiac contractility in settings of heart disease. 119 Accordingly, knockdown of PLN or increasing PLN phosphorylation might be promising therapeutic approaches to increase SERCA2a activity in treating heart failure. 120
Antisense RNA, small interfering RNA (siRNA) and short hairpin RNA (shRNA), are commonly used to specifically down regulate PLN expression and reverse the dysfunction of cardiac contraction of animal models in heart failure.121–124 Deletion of PLN using adeno-viral gene transfer leads to enhanced SERCA2a function and rescuing rats from ventricular failure. 121 However, complete ablation of PLN may cause lethal heart failure in human, which suggest the importance of PLN for human cardiac health in another way. 125 In this case, PLN mutants show great clinical prospects. It has been already confirmed that recombinant adeno-associated virus (rAAV)-mediated transcoronary administration of pseudophosphorylated mutant of human PLN (S16E) to hamster model of heart failure can enhance myocardial SR Ca2+ uptake and restore cardiac function. 126 Furthermore, the expression of Ad-S16E of PLN in sheep model of rapid ventricular pacing reverses the development of heart failure. 127 More recently, loss-of-function PLN mutants show sufficient affinity to displace the binding of PLN and activate SERCA. 128 Protein phosphatase 1 (PP1) inhibitors have been widely used to improve SR Ca2+ uptake due to the increasing phosphorylation of PLN. Improved cardiac performance is achieved by gene therapy vectors that inhibit PP1 activity in animals.129,130 Intracoronary delivery of I-1c (endogenous inhibitor against PP1) via adeno-associated viral-9 (AAV9) to pigs with failing heart preserves cardiac function and reduces the scar size caused by myocardial infarction. 129 Similarly, AAV9-mediated shRNA gene transfer specifically suppresses the expression of PP1β subunit, which significantly augments PLN phosphorylation and relieves cardiac diastolic function and remodelling. 130
Several drugs are also widely applied to attenuate the inhibition of PLN. It is reported that istaroxime and resveratrol can increase SERCA2a function by relieving PLN inhibition, which are utilized in improving cardiac function and lipopolysaccharide-induced cardiac dysfunction.131–133 Likewise, a pyridone derivative synthesized and discovered at Takeda Pharmaceutical Company attenuates PLN inhibition to SERCA2a, ultimately upgrading SERCA2a activity and enhancing the systolic and diastolic functions of the heart in rat. 134 Moreover, PLN-specific RNA aptamer, a PLN inhibitor with high affinity, is also reported to enhance both Ca2+transients and contractile function in normal rat cardiomyocytes, which has been considered as a novel therapeutic agent for improving heart failure instead of gene delivery by adenovirus. 135
Targeting SERCA (directly)
Apart from indirectly restoring SERCA2a function by targeting PLN, some other SERCA2a regulators previously acknowledged are also used to prevent the reduction or restore SERCA2a-mediated Ca2+ uptake in heart failure. For example, exogenous nitroxyl exerts positive inotropic and lusitropic effects in cardiomyocytes, which is due to increased S-glutathiolation at C674 of SERCA. 63 Overexpressed SUMO1 via gene transfer can maintain the amount of SERCA2a and significantly improve cardiac function in mice with heart failure. 71 β1-adrenergic receptor (AR) 136 and a small molecule (N106) 72 that can enhance SUMOylation of SERCA2a, result in significant improved contractility in vitro and in vivo. Moreover, pharmacological action and gene delivery have emerged as promising approaches to increase the expression and/or activity of SERCA2a, which have been developed as therapeutic strategies for rescuing cardiac contractile dysfunction in heart failure.
Pharmacological action
β-blockers, ACE inhibitors and aldosterone antagonists are the first-choice drugs applied in the treatment of cardiac diseases, which may prolong life and relieve the symptoms. To our regret, they cannot correct the underlying causes of contractile dysfunction. 137 Even though, from then on, drugs used individually or in combination have significant benefits to retain SERCA2a abundance, thereby improve contractility in heart failure, and it is extremely attractive at present. Among these drugs, by blockading the renin–angiotensin system, an angiotensin-converting enzyme inhibitor – enalapril – and an angiotensin II receptor antagonist – losartan – can partially prevent the down-regulation of SERCA2 protein and mRNA content as well as reduction of left ventricular function in a rat model of congestive heart failure. 138 Resveratrol, a natural antioxidant agent, utilized in a variety of diseases including heart diseases, is also found to lift SERCA2a expression and cardiac function by activating SIRT1 in streptozotocin-induced diabetic mice. 139 Likewise, insulin-like growth factor 1 (IGF-1) treatment is able to regain the amount of SERCA2a in diabetic rat cardiomyocytes, which is mediated by activation of the PI3kinase-Akt-SERCA2a signalling pathway. 140 Recently, some components extracted from plants reveal to exert cardioprotective effects by restoring SERCA2 expression. Oxymatrine, an anti-inflammatory agent extracted from the Chinese herb Sophora japonica, is able to improve cardiac function via upregulating SERCA2a expression in a rat model of chronic heart failure. 141
In addition, some other drugs that can modulate SERCA activity as indirect means to improve contractility in heart failure are reported. Glucocorticoid can prevent additional deterioration of SERCA2a activity in piglets with ischaemic cardiac dysfunction. 142 Hydralazine, a potential DNA methylation inhibitor, can increase SERCA function via decreasing methylation in the SERCA2a promoter, modulate Ca2+ handling and improve cardiac function in rats with isoproterenol-induced heart failure. 143 Istaroxime, a Na+/K+ ATPase inhibitor, can improve Ca2+ cycling, which is essential to the enhancement of Ca2+ uptake into SR, leading to increased SERCA function in cardiac SR vesicles of dogs with heart failure. 132 A clinical trial with the administration of istaroxime is demonstrated to improve pulmonary capillary wedge pressure and decrease heart rate in patients with heart failure via relieving the inhibition of PLN on SERCA2a. 133 In addition, CDN1163, a potent allosteric activator of SERCA, directly binds SERCA, alleviates ER stress and shows potential as a therapeutic for Parkinson’s disease, 144 type-2 diabetes and metabolic dysfunction. 145 However, its functions in heart has not yet been reported. Collectively, these studies show that pharmacological activation of SERCA2a activity may ameliorate several of the underlying dysfunctions associated with heart failure.
Gene therapy
The application of gene transfer technology to rescuing cardiovascular disease has been in development for over 20 years. Adenoviral (AV) expression system is utilized to directly increase SERCA expression for the first time in ventricular cardiomyocytes from patients with end-stage heart failure. 146 In this study, overexpressed SERCA2a by AV gene transfer generates increased contractility and accelerated relaxation, which suggests that gene-based therapy in human heart failure may offer new possible treatment strategy of this disease. Subsequently, many studies further verify the beneficial effects of SERCA2a gene transfer on the improvement of cardiac performance in experimental animals of heart failure,147,148 and increasing number of viral vectors are used in treatment with cardiac disease. Currently, recombinant viral vectors have the highest transfection efficiency and increased long-term transgenic expression, which have put them under the spotlight. 149 Among many viral vector systems examined so far, AV and adeno-associated virus (AAV) are the most commonly used vectors in cardiovascular disease.
Recombinant AV vectors are capable of transferring various genes to animal models, which have been extensively used in transferring gene to all major myocardial cell types in vitro and in vivo. With the ease of their production and the broad cell tropism, AVs are often available in pre-clinical gene therapy studies and cardiovascular gene therapy clinical projects. Rats injected with Ad-SERCA2a, for example, can normalize the decreased SERCA expression and attenuation of ATPase activity and even restore the ventricular function in rats with hypertrophy and heart failure. 150 Another group demonstrates that gene therapy using the Ad-SERCA2a is associated with the improvement in cardiac function in rats with experimental heart failure. 151 Moreover, overexpression of SERCA2a in failing human cardiomyocytes results in a restoration of SERCA protein and activity to non-failing level, which would provide a new insight into treatment of patients with heart failure using gene transfer technology. 146 However, expression of AV-delivered transgenes is short-lived and highly immunogenic, which is likely a major reason for their severely declining application in clinical settings.149,152 To avoid these problems, AAV is considered superior to AV in ongoing gene therapy trial for the treatment of cardiovascular disease, which is prone to contribute to their low immunogenicity and high efficiency. 149
In pigs with volume-overload heart failure, long-term overexpression of SERCA2a is achieved by rAAV1-mediated intracoronary gene transfer, in which preserved systolic function and improved ventricular remodelling is observed. 153 Likewise, left ventricular function is significantly improved in the AAV2/1-SERCA2a-treated sheep with rapid-pacing-induced heart failure. 154 An association between SERCA2a overexpression and reduced frequency of arrhythmias is observed using recombinant AAV vectors in rodents with chronic heart failure, 155 suggesting that SERCA2a has positive inotropic and lusitropic effects, and SERCA2a gene therapy, therefore, has anti-arrhythmic effects and may be a promising strategy for heart failure. Despite many studies have shown great potentials of gene transfer in animal models of heart failure, few can been translated to randomized clinical trials for the devastating disease.
Based on these findings, some randomized, placebo-controlled clinical trials using gene transfer to improve the function of the failing heart in patients are performed. 156 Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID), the first phase 1/2 clinical trial of AAV1 carrying SERCA2a in patients with advanced heart failure, was administered in 2007. Phase I portion is open-label and sequential dose escalation, during which no unexpected safety concerns is observed and several patients show improvements in relevant parameters, which demonstrates the safety and efficacy.157,158 On the contrary, phase II is a double-blind placebo-controlled randomized study.157,159 In this work, 39 patients with advanced heart failure are involved, who receive either placebo or one of three doses of AAV1-SERCA2. At up to 12 months of follow-up, significant increases in time to clinical events and decreased frequency of cardiovascular events are observed, and the average duration of cardiovascular hospitalizations is decreased for patients with high-dose treatment versus placebo. Remarkably, the long-term (3 year) CUPID via intracoronary infusion of AAV1/SERCA2a in patients with advanced heart failure is further performed. 160 The results show positive signals of cardiovascular events (including myocardial infarction, worsening heart failure, heart failure-related hospitalization, ventricular assist device placement, cardiac transplantation and death) and there is no safety concerns during the tracked 3 year. Despite of the small number of enrolled patients in this work, the favourable effects seen in CUPID still need to be replicated and validated in larger groups of patients. More recently,161,162 250 patients with class II–IV symptoms of heart failure are randomly assigned (1:1), who receive intracoronary infusion either placebo or AAV1-SERCA2a (1 × 10¹³). After up to 12 months of follow-up, no significant improved outcomes are obtained at the dose of AAV1/SERCA2a tested, which can stimulate further research and help to inform the design of future gene therapy trials.
Conclusion and perspectives
Heart failure remains a major health problem in western world. Despite advances in treatment have been achieved, high morbidity and mortality remain unacceptably. Over the last two decades, significant progress has been made in designing new therapeutic approaches for heart failure. Accumulating evidence proves that aberrant SR Ca2+ handling in cardiomyocytes is the primary defect, which is contributed in great degree by SERCA2a, a versatile Ca2+ transporter. Altered expression or function of SERCA2a is observed in many heart diseases, and correcting such aberration favourably affects Ca2+ flux and improves cardiomyocytes function, which has been widely accepted as a promising strategy in cardiac dysfunction treatment. Collectively, targeting PLN to indirectly retain the activity of SERCA2a or ratio of PLN to SERCA2a and increasing SERCA2a content and activity via drugs or gene delivery are the most promising therapies.
Although growing evidence shows that targeting SERCA2a is an useful approach in curing heart failure, whereas a large number of potential obstacles still exist. Not least of these is the choice of AAV vectors, which is the most advantagous method for treating failing heart, and it has been successfully stepped into phase I and II clinical trials, which is currently in phase 3 clinical study. AAV-mediated gene delivery has achieved with reduced inflammatory response, but two major problems need to be solved: usage of neutralizing antibodies against AAV precludes their effective use and relatively low transduction efficiency. In addition, long-term effects need to be put an effort in larger number of patients. Hopefully, with the realization of current trials targeting SERCA2a function in heart failure, alternative approaches to increase the efficacy of the therapy, such as using transplantation of induced pluripotent stem cell163,164 or deriving de novo cardiomyocytes, 165 may lead to enhanced long-term benefit to all the patients. It is clear that much more endeavour is necessary when we moved from bedside to bench and back again.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant no. 81700237, 31571172); Chongqing Research Program of Basic Research and Frontier Technology grant cstc2016jcyjA0407; China Postdoctoral Science Foundation (grant no. 2016M592623); Chongqing Postdoctoral Science Foundation (grant no. Xm2015033) and Fundamental Research Funds for the Central Universities (grant no. 0236015202008).