
Abstract
To study whether hypercaloric diet–induced obesity deteriorates vascular contractility of rat aorta through functional changes in α1 adrenergic and/or AT1 Angiotensin II receptors. Angiotensin II- or phenylephrine-induced contraction was tested on isolated aorta rings with and without endothelium from female Wistar rats fed for 7 weeks with hypercaloric diet or standard diet. Vascular expression of Angiotensin II Receptor type 1 (AT1R), Angiotensin II Receptor type 2 (AT2R), Cyclooxygenase-1 (COX-1), Cyclooxygenase-2 (COX-2), inducible Nitric Oxide Synthase (iNOS) and endothelial Nitric Oxide Synthase (eNOS), as well as blood pressure, glucose, insulin and angiotensin II blood levels were measured. Diet-induced obesity did not significantly change agonist-induced contractions (Emax and pD2 hypercaloric diet vs standard diet n.s.d.) of both intact (e+) or endothelium free (e−) vessels but significantly decrease both phenylephrine and angiotensin II contraction (Emax p < 0.01 hypercaloric diet vs standard diet) in the presence of both prazosin and losartan but only in endothelium-intact vessels. Diet-induced obesity did not change angiotensin II AT1, AT2 receptor proteins expression but reduced COX-1 and NOS2 (p < 0.05 vs standard diet). Seven-week hypercaloric diet–induced obesity produces alterations in vascular adrenergic and angiotensin II receptor dynamics that suggest an endothelium-dependent adrenergic/angiotensin II crosstalk. These changes reflect early-stage vascular responses to obesity.
Keywords
Introduction
There has been an epidemic increase in obesity [World Health Organization (WHO)] as a result of hypercaloric diet (HD) intake and sedentary habits. Both obesity and overweight are conditions associated with metabolic dysfunction, a decreased insulin sensitivity and diabetes. On the other hand, insulin resistance (IR) is known to be a key factor for the development of vascular damage such as endothelial dysfunction and impaired vascular relaxation. 1
Both renin–angiotensin system (RAS) and adrenergic system play a critical role in the control of physiological and pathological cardiorenal functions. 2 Indeed, Angiotensin II (Ang II) induced vasoconstriction and remodelling through AT1R has been involved in the pathogenesis of diabetic vascular dysfunction. 3
Then, under diabetic/obesity conditions, endothelium-derived vasodilators may increase their participation as a counter-regulatory mechanism towards homeostasis including an increased participation vasodilator components of RAS as AT2R 4 or angiotensin-converting enzyme 2 (ACE2)-angiotensin 1–7.5–7
Additionally, damage produced by metabolic disturbances can change adrenergic or Ang II receptors functionality, increasing the crosstalk between α1 and AT1 receptors.8–11 Indeed, an increased α1/AT1 crosstalk has been proposed as an early damage indicator of metabolic disturbances. 12
Obesity-induced cardiovascular and metabolic changes have been studied mainly in male animal models using high fat 13 or fructose diet intake,14,15 which have shown an increased metabolic sensitivity with respect to females.16,17 So reports about the cardiovascular impact of HD in female rodents are needed in order to clearly establish the possible intergeneric differences.
Considering aorta Ang II response has been used as a surrogate measure of large artery disease, 12 in this work, we, therefore, intended to study if HD for 7 weeks deteriorate vascular contractility of female rat aorta through functional changes in α1 and/or AT1 receptors and the role that endothelium may have in these changes.
Methods
Twelve-week-old female Wistar rats weighing 250 ± 15 g were used. Animals were kept in 12 h light/dark cycle and controlled humidity, with free access to food and water. All procedures were followed according by the Official Mexican Norm (NOM-062-ZOO-1969) for the Animal Handling and were approved by the local Ethical Committee of our institution Escuela Superior de Medicina, Instituto Politécnico Nacional.
Diet-induced obesity
Rats had free access to a standard diet rat chow (SD) (3.1 kcal/g) or to a HD (6.3 kcal/g) over 7 weeks. HD was prepared with 33% ground commercial rat chow, 33% full fat sweetened condensed milk (Nestle), 7% sucrose and 27% water. The diets and the water were provided ad libitum. Weight was recorded weekly. 18
At the end of this period, glucose tolerance test (GTT) as well as plasma levels of insulin and Ang II were determined and the aortas excised for molecular determinations.
Records in whole animal
Measurement of blood pressure (BP) was performed by indirect tail cuff plethysmography method (Letica 5007 PanLab, Barcelona). Rats were subjected to previous training for 3 days. Measurements were obtained at the beginning of the experiment and at the end of weeks 4 and 7. Procedure consisted in placing rats into appropriate traps inside a room free from noise and light, previously warmth to 32°C. Systolic BP was determined as the mean value after three consecutive successful measurements.
A drop of blood obtained from the tail tip was used to determine blood glucose levels with an Accu-Chek Advantage glucose meter (Roche Diagnostics, Basel, Switzerland).
GTT
After an overnight fast, baseline glucose was measured (0 min) and 5, 10, 15, 30, 45, 60, 90 and 120 min subsequently to an oral bolus of 1 g/kg glucose.
Studies in isolated organ
Under ether anaesthesia, animals were sacrificed and the thoracic aorta was excised and cut into ring segments (3–4 mm long). Endothelium was removed from three of them. Tissues were suspended on wire hooks in isolated b tissue baths under 3 g tension containing 10 mL Krebs–Henseleit solution, maintained at 37°C, pH 7.4 and bubbled with 95% O2/5% CO2. After a 90-min equilibration period, isometric responses were recorded using a 50G-TSD125C force transducer, an amplifier DA100C and a data acquisition system MP100 (Biopac System Inc., Santa Barbara, CA, USA). Viability of aortas was evaluated with KCL 80 mM, and to assess the presence of endothelium, vessels were contracted with phenylephrine (Phe 1 × 10−6 M) and relaxed with acetylcholine (Ach 10−6 M). Rings were considered to have endothelium if relaxation was ⩾80%. Cumulative concentration–response curves to Ang II (10−5 to 10−10 M) and Phe (10−4 to 10−10) were done in vascular rings both with endothelium (e+) and without endothelium (e−) (n = 5). Some curves were ran in the presence of AT1R-competitive antagonist losartan (1 × 10−8 M) or alpha1 selective adrenergic antagonist prazosin (3.1 × 10−10 M). Concentrations were chosen considering Ki for vascular tissue (16.2 ± 4.6 nm or 0.04–0.08 nM) for losartan or prazosin, respectively.19,20
Graphs were constructed using the percentage of contraction with respect to KCl maximal effect (100%).
Determination of proteins by Western blot
Protein expression of eNOS, iNOS, COX-1 and COX-2 enzymes and of AT1 and AT2 receptors was determined through Western blot (WB) technique. Intact vessels cleaned of connective tissue (n = 4 per group) were homogenized in Radioimmunoprecipitation assay (RIPA) buffer containing a mixture of protease inhibitors at low speed (between 10,000 and 15,000 r/min during 15 s for each pulse) followed by 10,000 r/min for 10 min at 4°C centrifugation. Protein concentration was determined with the Lowry method. After 2-mercaptoethanol (100°C for 10 min) treatment, equal amounts of protein (50 mg) were loaded on a 10% and 5% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). They were subjected to electrophoresis (MiniPROTEAN®) 25 min to 80 V and 1.25 min to 120 V and transferred to polyvinylidene fluoride membranes for 1 h at 15 V, using a semi-dry trans blot (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked 2 h at room temperature in 5% low-fat milk washing solution. Then, membranes were incubated with goat polyclonal antibody (Santa Cruz Biotechnology, CA, USA) against AT1R, AT2R, COX-1,COX-2, actin or rabbit polyclonal antibody against iNOS and eNOS diluted 1:200, 1:400 and 1:1000, in washing solution at 4°C overnight.
Membranes were then washed five times, incubated with rabbit anti-goat or goat anti-rabbit horseradish peroxidase-conjugated second antibody 1:10,000 for 2 h at room temperature and washed extensively. Membranes were incubated with chemiluminescence blotting substrate (Western Blotting Luminol Reagent; Santa Cruz Biotechnology) according to the manufacturer’s protocol and exposed to film that was immediately developed. The film was scanned and band intensity was measured by computer analysis, using gel densitometer BioSens SC 645 and was normalized with actin intensity (control protein).
Blood sampling
At the end of the seventh week, and after overnight fasting, animals were sacrificed and blood samples were obtained via cardiac puncture. Samples were stored at 4°C in Eppendorf tubes containing heparin. They were centrifuged right after to 1500 r/min for 15 min to 4°C. One millilitre of serum was removed and 100 mL of protease inhibitor mixture was added. Immediately, the serum was frozen at −70°C for analysis of plasma peptide C and Ang II concentrations.
Determination of plasma angiotensin II and peptide C
The determination of plasma levels of angiotensin II and peptide C – as a measure of insulin concentration – was conducted by ELISA kit for Ang II (Angiotensin II EIA kit, Cayman®) or ELISA kit for peptide C (Human C-peptide ELISA, Millipore®) following the manufacturer’s recommendations.
Analysis and statistics
In isolated organ experiments, each experimental group included five to six animals. Data are expressed as the mean ± SEM. pD2 (−Log EC50) and Emax values were obtained by non-linear regression analysis from concentration–response curves. Statistical evaluation of the data, when comparing each point of concentration–response curve, was carried out by two-way analysis of variance (ANOVA), with Bonferroni test for comparison of means. pD2 (−Log EC50) and Emax values were compared using unpaired Student’s t test.
For the WB, values are expressed as arbitrary units that result from the coefficient AT1 or any/actin. They are the mean ± SEM of four experiments and were analysed using unpaired Student’s t test. In all comparisons, values of p < 0.05 were considered to indicate significant differences between the means.
Results
After 7 weeks, HD significantly increased weight compared with SD rats [15.15 ± 2.96 (SD) vs 37.39 ± 4.16 (HD) g; p < 0.05]. Interestingly, BP was unchanged in HD animals (140 ± 3.521 vs 133 ± 2.65 mm Hg HD vs SD, respectively, n.s.d.); blood glucose increased in HD with respect to SD (113.75 ± 0.75 vs 87 ± 7.56 mg/dL; p < 0.05), as well as plasma Ang II levels (107.8 ± 8.59 vs 82.31 ± 7.08 ng/mL HD vs SD, respectively; p < 0.05). However, plasma insulin levels (0.92 ± 0.12 vs 0.79 ± 0.09 ng/mL HD vs SD, respectively; n.s.d.) and homeostatic model assessment (HOMA) index (6.63 ± 1.11 vs 5.95 ± 1.38 HD vs SD, respectively; n.s.d.) were unchanged (Table 1).Also, no difference between the experimental groups was found in the GTT (14,570 ± 996 vs 16,720 ± 517.8 AU SD and HD, respectively; n.s.d.).
Biochemical parameters.
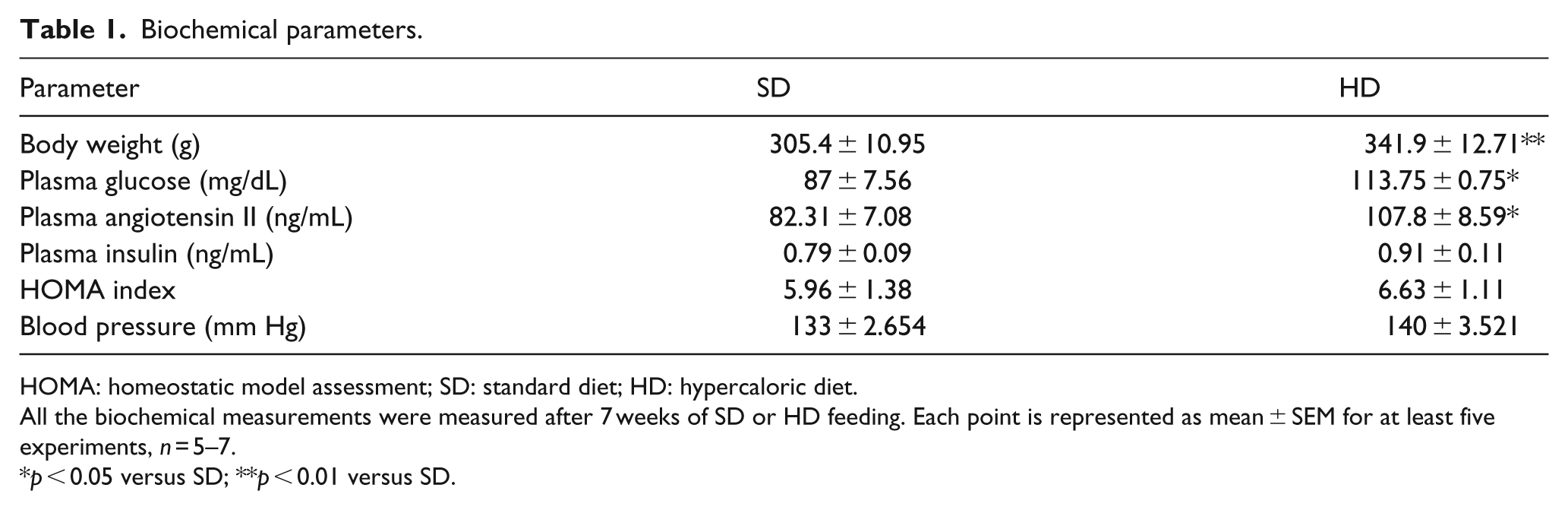
HOMA: homeostatic model assessment; SD: standard diet; HD: hypercaloric diet.
All the biochemical measurements were measured after 7 weeks of SD or HD feeding. Each point is represented as mean ± SEM for at least five experiments, n = 5–7.
p < 0.05 versus SD; **p < 0.01 versus SD.
Response to angiotensin II
In order to determine whether HD changed Ang II-induced contraction, concentration–response curves to Ang II were ran in aorta rings both with endothelium and without endothelium (n = 5) in both experimental groups. To evaluate the role of AT2 receptors, some curves were ran in the presence of losartan (1 × 10−8 M). Crosstalk was evaluated testing the effect of prazosin (3.1 × 10−10 M).
Endothelium intact (e+)
HD did not change aorta contraction to Ang II (Emax 10.27% ± 1.99% vs 8.24% ± 2.18% SD and HD, respectively; n.s.d.) (Table 2). Incubation with losartan reduced the contraction of HD vessels (Emax 8.4% ± 1.04% vs 3.1% ± 0.81%; p < 0.05, SD and HD, respectively). The same decrease was observed with prazosin incubation (Emax 7.7% ± 0.44% vs 2.49% ± 0.45%; p < 0.05, SD and HD, respectively) (Table 2, Figure 1(a) and (c)).
Angiotensin II Emax (%) and pD2 (log M) developed by aortic rings with and without endothelium from rats fed SD or HD.
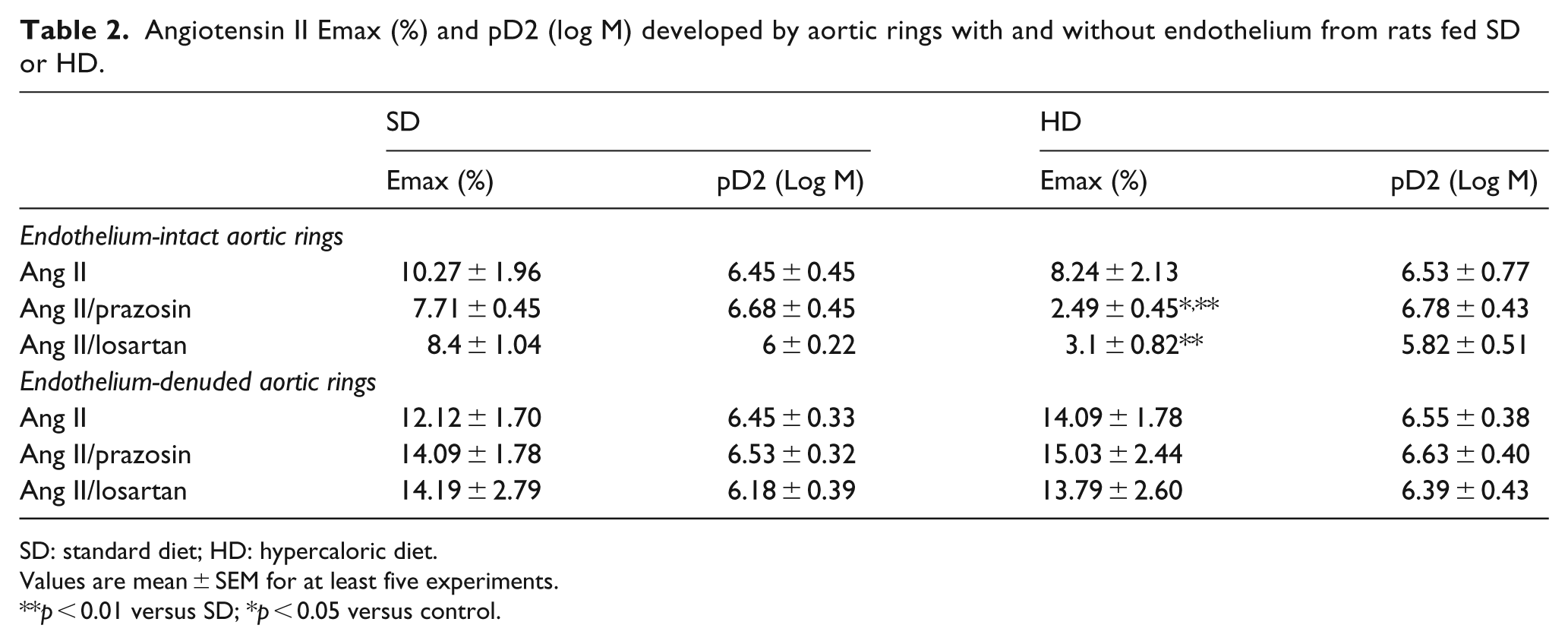
SD: standard diet; HD: hypercaloric diet.
Values are mean ± SEM for at least five experiments.
p < 0.01 versus SD; *p < 0.05 versus control.
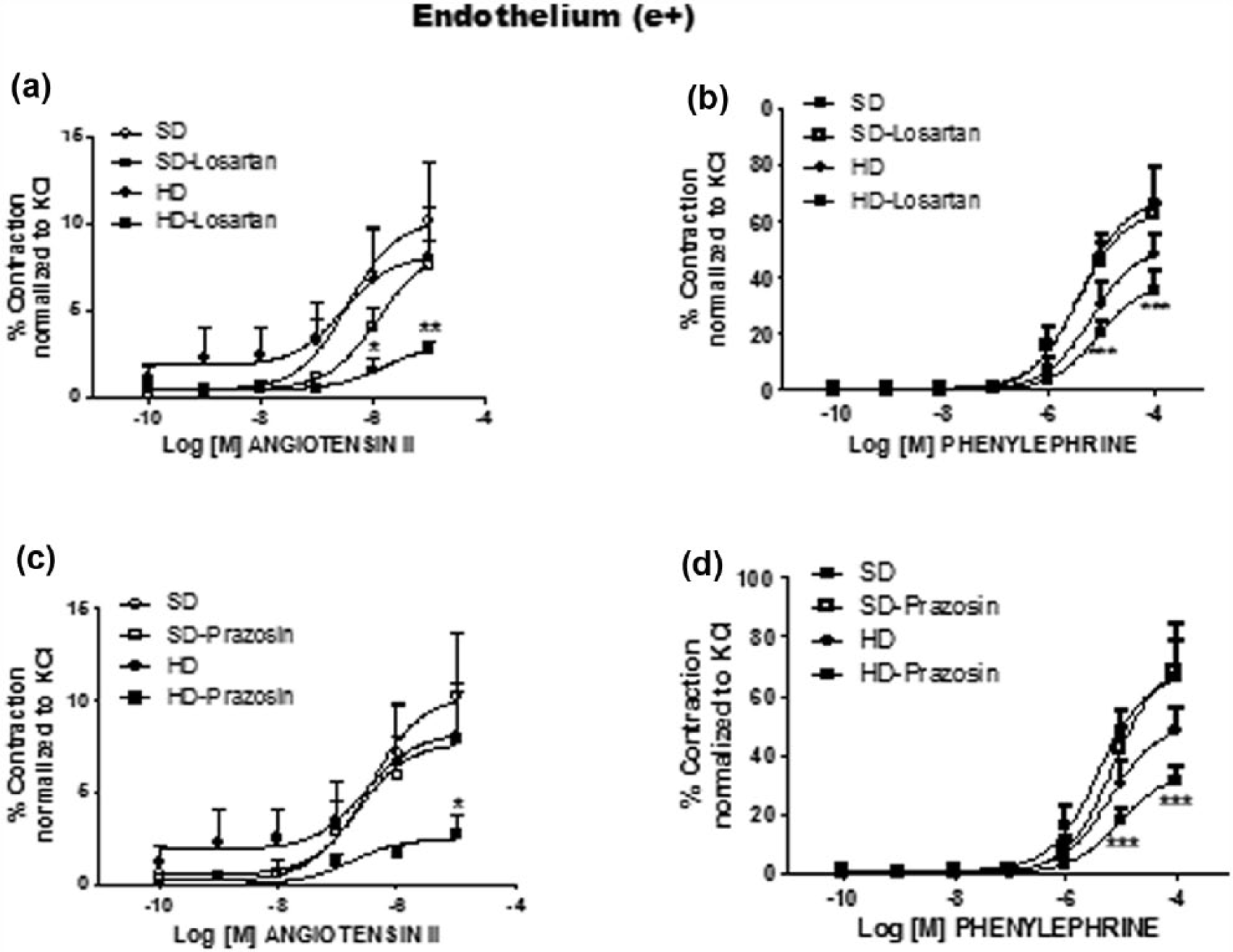
Effect of hypercaloric diet (HD) on the (a and c) angiotensin II or (b and d) phenylephrine cumulative concentration–response curve on thoracic aortic rings with endothelium: rats fed standard diet (SD) control (○), in the presence of losartan (1 × 10−8 M) (a and b) or prazosin (3.1 × 10−10) (c and d) (□); rats fed with HD control (●), in the presence of losartan (1 × 10−8 M) (a and b) or prazosin (3.1 × 10−10) (c and d). p < 0.05 versus SD, **p < 0.01 versus SD, ***p < 0.001 versus SD.
Endothelium-denuded rings (e−)
HD did not affect Ang II-induced contraction of (e−) vessels (Emax 12.12% ± 1.70% vs 14.09% ± 1.78%; n.s.d.); the results showed no difference between SD and HD groups in the presence of losartan (Emax 14.19% ± 2.79% vs 13.79% ± 2.60% SD and HD, respectively) or prazosin (Emax 14.09% ± 1.78% vs 15.03% ± 2.44% SD and HD, respectively; n.s.d.) (Table 2, Figure 2(a) and (c)).
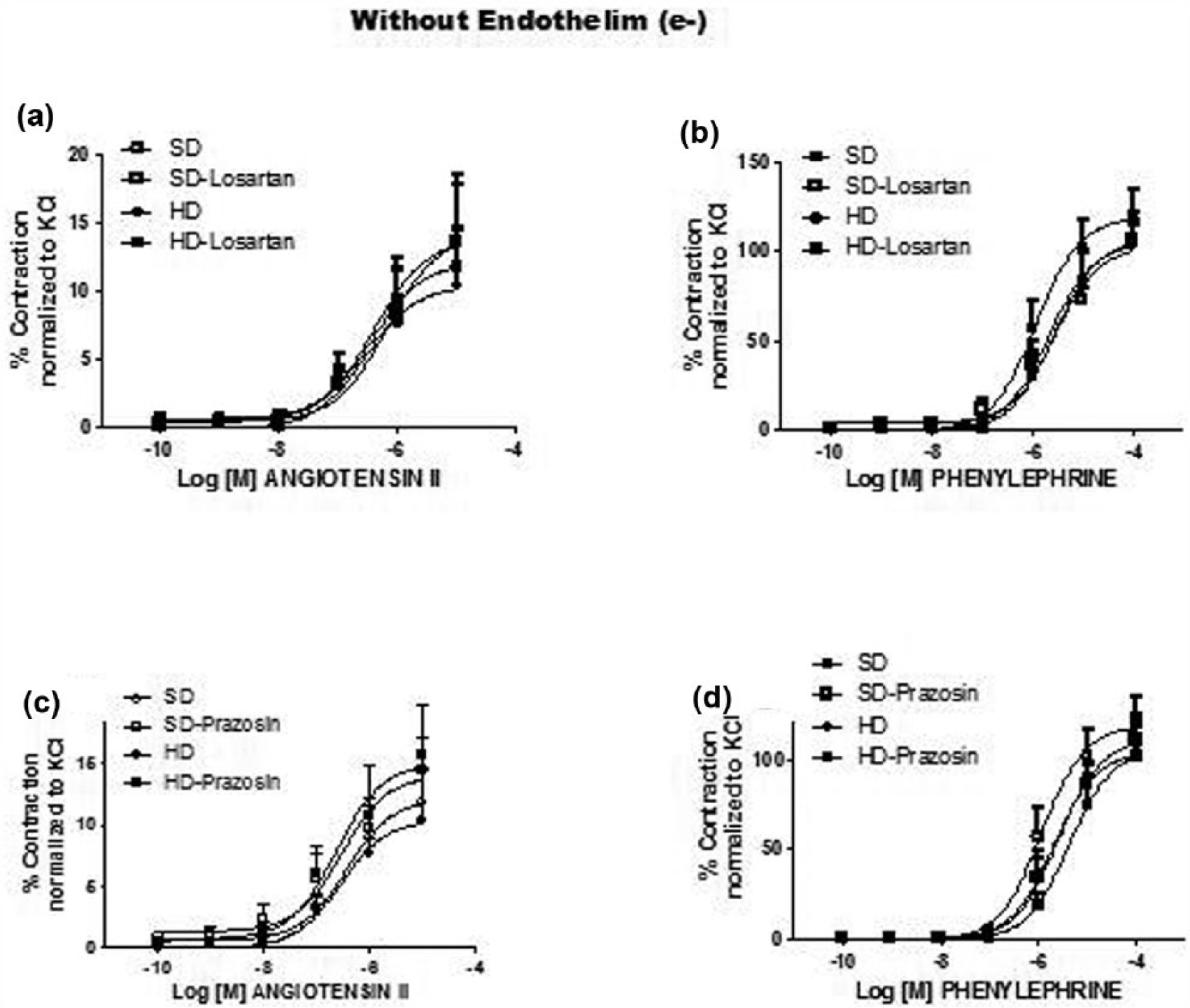
Effect of hypercaloric diet (HD) on the (a and c) angiotensin II or (b and d) phenylephrine cumulative concentration–response curve on thoracic aortic rings without endothelium: rats fed standard diet (SD) control (○), in the presence of losartan (1 × 10−8 M) (a and b) or prazosin (3.1 × 10−10) (c and d) (□); rats fed with HD control (●), in the presence of losartan (1 × 10−8 M) (a and b) or prazosin (3.1 × 10−10) (c and d). p < 0.05 versus SD, **p < 0.01 versus SD, ***p < 0.001 versus SD.
Response to phenylephrine
In order to determine whether the effect of hypercaloric diet-induced obesity was agonist specific, phenylephrine (Phe) contraction was evaluated (n = 5).
As with Ang II, HD did not change Phe contraction of (e+) aorta rings (Figure 1) (Emax 68.3% ± 6.58% vs 50.97% ± 5.15% SD and HD, respectively; n.s.d.) (Table 3), but decreased the response in ring incubation with losartan (Emax 63.87% ± 4.252% vs 38.8% ± 3.588%; p < 0.01; SD and HD respectively) or with prazosin (Emax 70.45% ± 7.13% vs 34.72% ± 3.04%; p < 0.01; SD and HD, respectively) (Table 3, Figure 1(b) and (d)).
Phenylephrine Emax (%) and pD2 (log M) developed by aorta rings with and without endothelium from rats fed SD and HD.
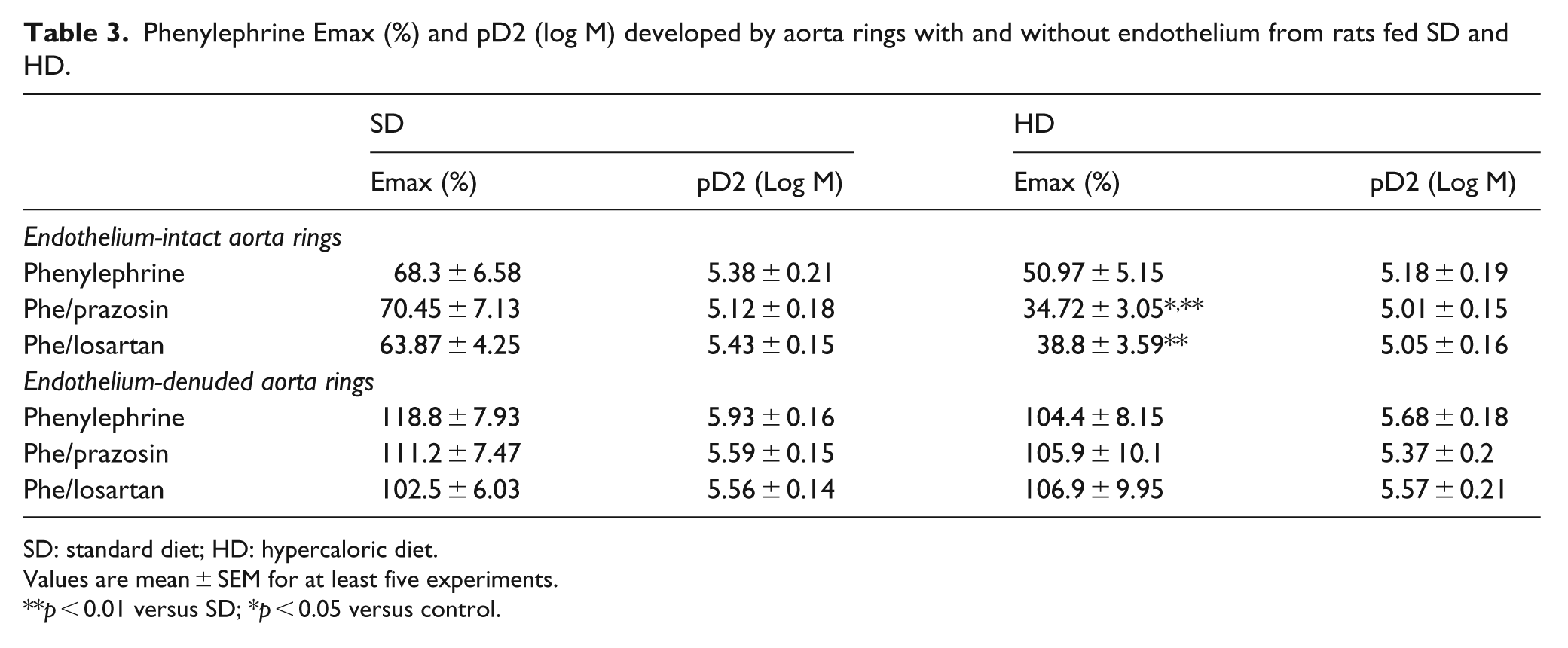
SD: standard diet; HD: hypercaloric diet.
Values are mean ± SEM for at least five experiments.
p < 0.01 versus SD; *p < 0.05 versus control.
Also, HD did not change Phe-induced contraction (Emax 118.8% ± 7.93% vs 104.4% ± 8.15% SD and HD, respectively; ; n.s.d.) (Table 3), nor in the presence of losartan (Emax 102.5% ± 6.03% vs 106.9% ± 9.95% SD and HD, respectively; n.s.d.) or prazosin (Emax 111.2% ± 7.47% vs 105.9% ± 10.1% SD and HD, respectively; n.s.d.) (Table 3, Figure 2(b) and (d)).
Protein expression
WB analysis showed no differences in the expression of Ang II, AT1 and AT2 receptors between the experimental groups (AT1R 0.5089 ± 0.06 vs 0.6722 ± 0.52 AU SD vs HD, respectively, n.s.d.; AT2R 0.3502 ± 0.06 vs 0.454 ± 0.07 AU SD vs HD, respectively; n.s.d.).
A significant decrease in COX-1 expression was found in vessels from HD compared with SD, while COX-2 was unchanged (COX-1 1.4 ± 0.51 vs 1.01 ± 0.13 AU SD vs HD, respectively, n.s.d.; COX-2 0.24 ± 0.04 vs 0.2 ± 0.14 AU SD vs HD, respectively; n.s.d.) (Figure 3).
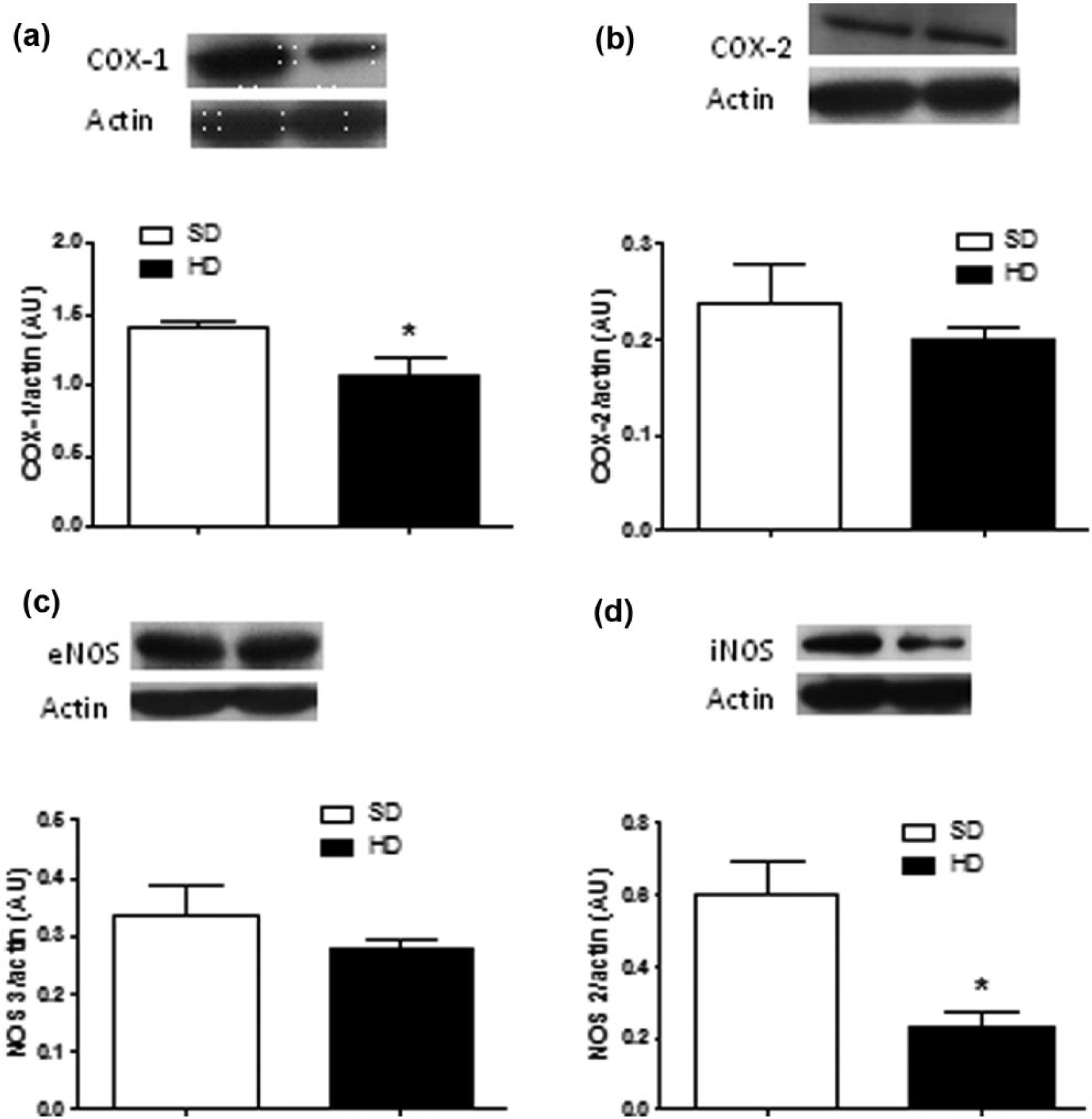
(a) Cyclooxygenase-I, (b) cyclooxygenase-2, (c) endothelial Nitric Oxide Synthase and (d) inducible Nitric Oxide Synthase from thoracic aorta. Rats fed standard diet (SD) (□) and hypercaloric diet (HD) (■) (COX-1: 1.4 ± 0.51 vs 1.01 ± 0.13 AU SD vs HD, respectively, n.s.d.; COX-2: 0.24 ± 0.04 vs 0.2 ± 0.14 AU SD vs HD, respectively, n.s.d.; eNOS: 0.33 ± 0.55 vs 0.27 ± 0.19 AU SD vs HD, respectively, n.s.d.; iNOS: 0.6 ± 0.09 vs 0.23 ± 0.44 AU; *p < 0.05). Data are the mean ± SEM of four rats.
Also, we found a lower iNOS enzyme protein expression in vessels from HD compared with SD, while eNOS was unchanged (eNOS 0.33 ± 0.55 vs 0.27 ± 0.19 AU SD vs HD, respectively, n.s.d.; iNOS 0.6 ± 0.09 vs 0.23 ± 0.44 AU *p < 0.05) (Figure 3).
Discussion
This study examined the effects of obesity induced with HD on angiotensin II or phenylephrine induced contractility and in the possible crosstalk between adrenergic alpha 1 and angiotensin II AT1 receptors.
Results showed 7-week HD increased weight and basal blood glucose values, which is in agreement with previous reported work using male rodents; 21 nevertheless, other metabolic indicators such as GTT, insulin levels or HOMA index were not impaired. Normal GTT, in contrast with the findings reported by Huan 21 who described HD induced an overt glucose intolerance, points out possible gender-based differences.
An increase in Ang II plasma levels as a result of HD was another interesting finding; these results confirm previous studies in which HD increases Ang II22,23 and support the relationship between obesity/IR and RAS, even when this increase was not enough to change AT1R and AT2R protein expression or systolic BP.
Seven-week HD did not change vasoconstriction, independently of the presence of endothelium. Previous studies in aorta of HD-fed C57BL/6 mice 24 and Sprague-Dawley rats 22 found an augmented Ang II response linked to increased AT1R expression as well as increased BP and vascular reactivity.3,14 Moreover, HD has demonstrated to rise BP 25 within a period of 4–8 weeks. 26
Differences with our findings can be attributed to the sex of the rodents. We used female rats (either stage of ovarian cycle), considering reports on female animals are scarce. Galipeau et al. reported that upon fructose feeding, female rats did not develop hypertension or hyperinsulinaemia, except after ovariectomy. 22 Besides, some studies suggest endothelial dysfunction and elevated BP in HD models depend on the presence of testosterone. 27 Then, our results support the idea female condition provides a resistance profile both for metabolic and cardiovascular impact of HD.
Ang II has been shown to play a role in HD and obesity-induced IR and vascular abnormalities.1,13 All RAS known components are present in blood vessels, and selective Ang II receptors stimulation may regulate vascular tone and, indirectly, insulin availability. Ang II, the more extensively studied and active peptide of the system, stimulates AT1R (mainly associated with vasoconstriction) and AT2R (associated with vasodilation). Vasodilation is also produced by mas receptors stimulated by angiotensin 1–7 (Ang 1–7) formed from Ang II by the ACE25 and by angiotensin IV derived from angiotensin III or Ang II 28 by mechanisms related to NO bioavailability.1,5
Ang II dose–response curves were ran blocking AT1R with losartan. Under these conditions, HD reduced Ang II response (Emax) in (e+) but not in (e−) thoracic aorta. A possible explanation of these results is that HD promotes Ang II stimulation of AT2R-NO endothelium-dependent vasodilation while AT1R are blocked. Accordingly, it has been described an enhanced relaxant response due to NO and coupling with ATP-sensitive K+ channels in diabetic vessels. 29 Also, previous reports in diabetic rat aorta described increased AT2 receptor expression 30 and Ang II induced relaxation in the presence, but not in the absence, of AT1 selective antagonists (losartan or valsartan). 29
To test the hypothesis that α1-adrenoceptors/angiotensin AT1 receptors crosstalk is enhanced in metabolic stressed vessels, 11 response of HD aortas to Ang II was evaluated in the presence of α1 antagonist prazosin. Blocking α1 receptors significantly decreased the contraction of (e+) but not of (e−) HD aorta rings. Given prazosin seems to be able to antagonize AT1R allowing Ang II stimulation of AT2R-NO endothelium-dependent vasodilation, these results can be understood as an increased α1/AT1 crosstalk that becomes evident under metabolic distress.
When contraction was induced with phenylephrine, HD per se did not change the response with respect to control regardless of endothelium. As with Ang II, Emax was reduced in HD vessels in the presence of both prazosin and losartan without change in apparent affinity (Kd). The decreased contractility observed in the presence of losartan suggests a α1/AT1 crosstalk.
The possibility that prazosin effect may evidence a change in the adrenergic receptors by HD is reduced, since α1D adrenoceptor protein expression, which is the main alpha adrenergic receptor described in the rat aorta 31 , remained unchanged (data not shown).
Then, considering α1-AT1R synergy in vasoconstriction, 4 results suggest that obesity induced by HD promote receptor changes that enable prazosin to antagonize AT1R, as was previously proposed 32 and to unmask endothelium vasodilators that counterbalance HD-induced pro-constrictors, such as endothelin-1 and AT1R-mediated oxidative stress. In agreement, prazosin has shown to antagonize AT1R in the same vessel 32 as well as to improve IR. Also, a crosstalk has been described in the smooth muscle of rabbit aorta10,11 that is modified by hypercholesterolaemia 12 and has been proposed as a mechanism for the onset and progression of chronic vascular diseases.
Ang 1–7 and mas receptor could also be involved, considering these factors have been reported increased in response to HD5,6 and ACE2 – the enzyme that converts Ang II to Ang 1–7 is regulated by a high-fat diet and high sucrose intake in rats.33,34 ACE levels were not measured in this work, but the hypothesis of increased participation of Ang 1–7 to explain the decreased response elicited by endothelium-intact HD aortas in the presence of antagonists cannot be discarded. Further research will examine whether HD-mediated enhancement of aortic endothelial function is mediated by the activation of ACE. Besides, while no significant change in the expression of eNOS and iNOS enzymes was found, COX-1 expression was decreased. The latter finding can be related to a decreased production of pro-vasoconstrictor prostaglandins such as PGH2 and TXA2 that act on thromboxane-prostanoid receptors. 35
Then, HD/obesity seems to trigger changes on vascular α1 and/or AT1 receptors sensitivity to antagonists provided Ang II or Phe-induced contraction were inhibited by prazosin and losartan, respectively. We add further evidence of an endothelium-dependent α1/AT1 receptors crosstalk under pro-diabetic conditions.
Also, the present data are compatible with an increased compensatory participation of endothelium vasodilators. The present model is useful to evaluate early vascular impact of obesity induced with HD. These early changes may be surmounted lately if the metabolic challenge continues for a longer period of time.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This study was partially supported by Consejo Nacional de Ciencia y Tecnología (CONACyT), Secretaría de Investigación y Posgrado, IPN (SIP) and Comisión de Operación y Fomento de Actividades Académicas, IPN (COFAA) grants.