
Abstract
Vascular complications contribute significantly to morbidity and mortality of diabetes mellitus. The primary cause of vascular complications in diabetes mellitus is hyperglycaemia, associated with endothelial dysfunction and impaired neovascularization. Circulating endothelial progenitor cells was shown to play important roles in vascular repair and promoting neovascularization. In this review, we will demonstrate the individual effect of high glucose on endothelial progenitor cells. Endothelial progenitor cells isolated from healthy subjects exposed to high glucose conditions or endothelial progenitor cells isolated from diabetic patients exhibit reduced number of endothelial cell colony forming units, impaired abilities of differentiation, proliferation, adhesion and migration, tubulization, secretion, mobilization and homing, whereas enhanced senescence. Increased production of reactive oxygen species by the mitochondria seems to play a crucial role in high glucose–induced endothelial progenitor cells deficit. Later, we will review the agents that might be used to alleviate dysfunction of endothelial progenitor cells induced by high glucose. The conclusions are that the relationship between hyperglycaemia and endothelial progenitor cells dysfunction is only beginning to be recognized, and future studies should pay more attention to the haemodynamic environment of endothelial progenitor cells and ageing factors to discover novel treatment agents.
Introduction
Vascular complications contribute significantly to morbidity and mortality of diabetes mellitus (DM). These complications were characterized by early development, bilateral involvement and rapid progression, due to atherosclerosis. 1 It has been estimated that 73% of adult diabetic patients are hypertensive and more than 65% die from cardiovascular disease or stroke. 2 Moreover, emerging evidence demonstrated that maternal diabetes has long-term effects on the offspring, 3 including the development of hypertension, elevated blood pressure 4 and neonatal hypoglycaemia.
The primary cause of vascular complications in DM is hyperglycaemia, associated with endothelial dysfunction and impaired neovascularization. 5 In fact, endothelial dysfunction has long been viewed as the first event for the development of atherosclerosis and clinical events. 6 Therefore, the maintenance of an intact endothelial layer and improving its function are essential for keeping vessels healthy and away from cardiovascular complications associated with diabetes. Neovascularization is necessary to restore the blood flow after myocardial ischaemia and atherosclerosis. There is increasing evidence that neovascularization has two cell sources. 7 One is from the proliferation and migration of the fully differentiated endothelial cells (ECs), a process referred to as angiogenesis. 8 The other is from the homing of the circulating progenitor cells [CPCs; endothelial progenitor cells (EPCs)] to sites of endothelial disruption and incorporating into nascent endothelium, 9 a process termed as vasculogenesis (Figure 1). It has been estimated in animal models that EPCs contribute to 25% of ECs in newly formed vessels. 10
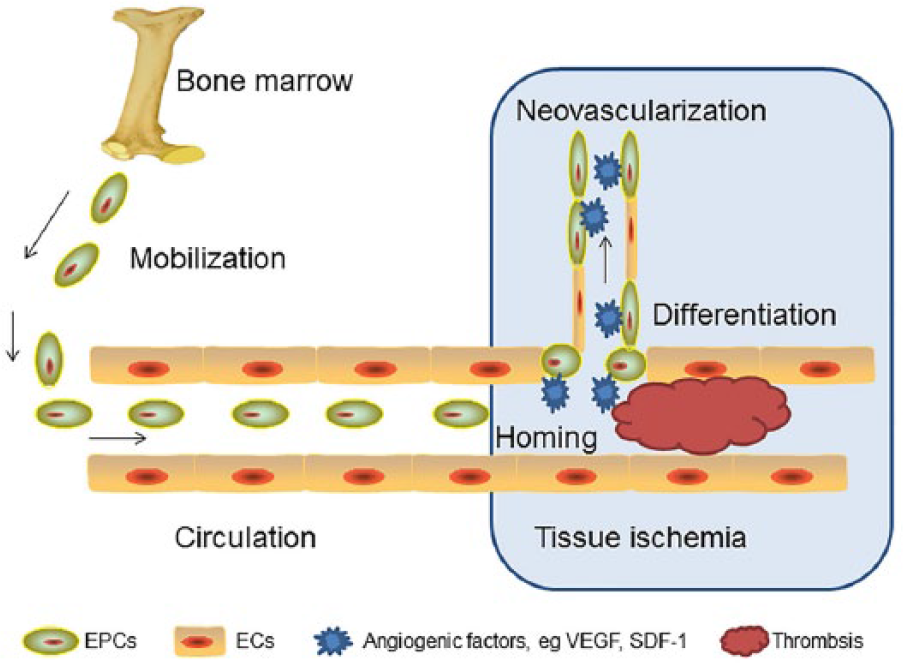
Schematic drawing of the EPCs participating in the process of neovascularization at the ischaemic tissue.
The EPCs were first isolated from the peripheral blood by Asahara et al. 7 Increasing evidence suggests that there are two types of EPCs, the so-called early and late EPCs. Early EPCs are spindle-shaped, characterized by the expression of CD14 and CD45, and show limited proliferating potential for long-term culture. The late EPCs display a cobblestone-like morphology, do not express CD14 or CD45, and have a higher proliferative potential.11,12 Compared with early EPCs, late EPC cultures seem more difficult to establish and sometimes impossible in some patients with DM and cardiovascular disease. 13 It has been demonstrated that EPCs are active players in maintaining a healthy cardiovascular system not only by integrating themselves into the newly developing capillaries but also by secreting the angiogenic growth factors in a paracrine manner. 14
Recently, there is increasing evidence showing the reduced number and impaired function of circulating EPCs induced by hyperglycaemia in diabetes. In this review, we will demonstrate the individual effect of high glucose on EPCs, including their morphology, differentiation, proliferation, adhesion, migration, tubulization, senescence, secretion, mobilization and homing. Other DM complications including diabetic nephropathy, retinopathy and cardiomyocyte associated with EPC dysfunction has been described elsewhere15,16 and will not be the focus of this review. In the later sections of review, we will discuss possible mechanisms underlying the impaired function of EPCs and the limited options currently available for alleviating EPC dysfunction exposed to high glucose conditions.
High glucose–induced EPC dysfunction
Clinical evidence
Emerging evidence has shown that the reduced number and impaired function of circulating EPCs are involved in vascular complications in diabetes (Table 1). Fadini et al. 34 quantified the CPCs (CD34+) and EPCs [CD34+ kinase-domain insert containing receptor (KDR)+] in 51 type 2 diabetic patients and 17 control subjects and found that the CPCs and EPCs were reduced by 33% and 40%, respectively, compared with healthy subjects. Furthermore, the diabetic patients with peripheral vascular disease were associated with a 47% reduction in EPCs, which for the first time, implied the involvement of depleted EPCs in the pathogenesis of peripheral vascular complications of diabetic mellitus. More recently, Balestrieri et al. 35 performed the EPC counts from 48 type 2 diabetic patients and 20 control individuals. In agreement with previous studies, they found the level of CD34 + KDR + cells was significantly reduced in type 2 diabetic patients compared with the healthy controls. Interestingly, this phenomenon was mediated by a platelet-activating factor (PAF)–silent information regulator 1 (SIRT1) pathway. The evidence was that SIRT1 protein levels were decreased while PAF receptors were upregulated in type 2 diabetic patients. This in vivo results were then confirmed by the in vitro experiments that EPCs stimulated with PAF accompanied by lower SIRT1 protein and messenger ribonucleic acid (mRNA) levels. These findings unveil the relationship between PAF and SIRT1 pathways in EPCs during altered glucose haemostasis. As CPCs and EPCs are associated with many aspects of cardiovascular disease, their levels show prognostic roles in general cardiovascular outcomes and death, 17 microangiopathy 18 and macroangiopathy 19 in type 2 diabetic patients.
High glucose–induced EPC dysfunction.
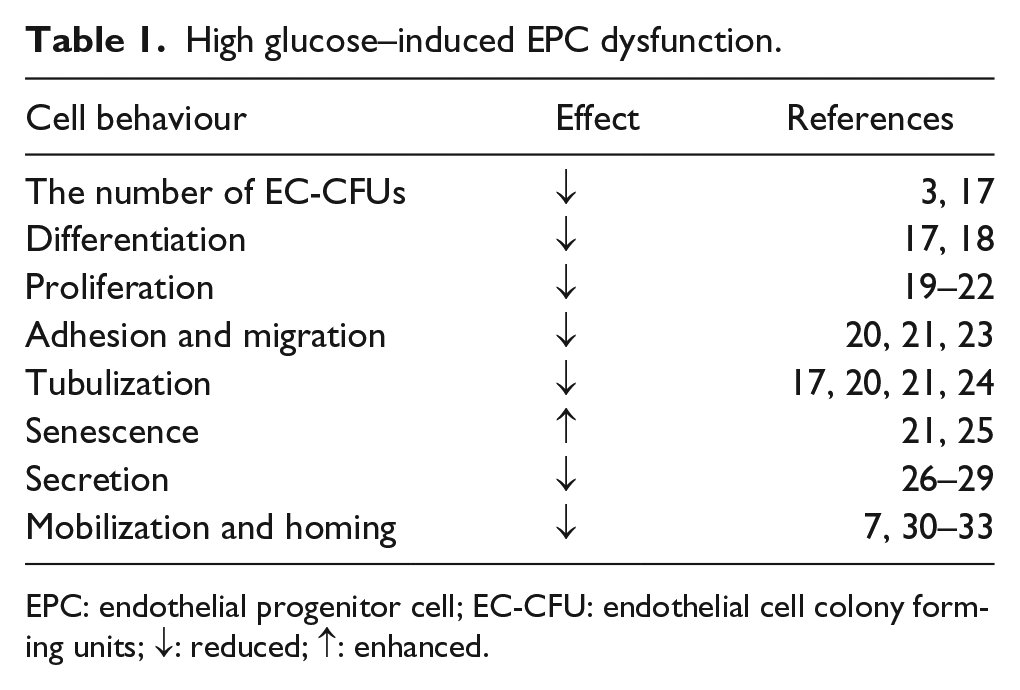
EPC: endothelial progenitor cell; EC-CFU: endothelial cell colony forming units; ↓: reduced; ↑: enhanced.
Moreover, the reduced EPC numbers and angiogenicity were also observed in type 1 diabetic patients. Type 1 diabetes is associated with reduced vascular repair, as indicated by impaired wound healing and reduced collateral formation in ischaemia. Loomans et al. 20 explored the EPC number and function in type 1 diabetes. They showed a 44% lower of the EPC number from type 1 diabetic patients relative to the age- and sex-matched control subjects. Furthermore, the angiogenesis ability of these patient EPCs were impaired, as tested its secretion of angiogenic growth factors using a 4-day EPC conditioned media in an in vitro angiogenesis model. These results provided evidence that EPC dysfunction contributes to the pathogenesis of vascular complications in type 1 diabetes.
Morphological analysis (the number of endothelial cell colony forming units)
EPCs cultured on fibronectin for 7 days can form colony unit [colony forming units (CFU)], with thin, flat cells emanating from a central cluster of spherical cells. However, when EPCs, isolated from peripheral mononuclear cells from healthy subjects, were cultured in high glucose medium (33 mM), the number of endothelial cell colony forming units (EC-CFU) was significantly decreased and the cells remained round/polygonal. 21 In order to uncover the underlying mechanisms reducing EPC CFUs when exposed to high glucose environment, Ingram et al. 3 evaluated the colony formation ability of EPCs isolated from human umbilical cord blood samples exposed to a wide range of hyperglycaemia observed in pregnant women with diabetes. The results showed that colony formation was reduced at 10, 15 and 30 mM dextrose concentrations compared with normal glycaemic condition (5-mM dextrose). Moreover, enhanced senescence, not apoptosis, contributes to this hyperglycaemia-induced reduction of colony formation of EPCs.
Differentiation
Cultured EPCs can differentiate into mature ECs with the expression of EC markers like VEGFR2 and CD34 to take part in the vascular repair. High glucose (33 mM) incubation of EPCs isolated from peripheral mononuclear cells from healthy subjects will inhibit their differentiation by reducing the number of DiI-acLDL and lectin-positive staining cells and downregulating their expression of VEGFR2 and CD31. This biological event was mediated by an Akt/forkhead box O transcription factors 1 (FoxO1) pathway and could be reversed by benfotiamine administration. 21 In addition, dimethylarginine dimethylaminohydrolase (DDAH) may also play a pivotal role. It has been demonstrated that the expression level of DDAH2 was increased with EPC differentiation, and interruption of DDAH2 expression inhibited vascular endothelial growth factor (VEGF) and KDR expression, while had no effect on asymmetric dimethylarginine (ADMA). 22 In type 2 diabetic patients, DDAH2 was shown to be downregulated in circulating EPCs, indicating high glucose–regulated EPC differentiation via a DDAH2-VEGF2/KDR pathway.
Proliferation
A major barrier to the clinical usage of EPCs as an autologous cell therapy product for neovascularization is their paucity in the peripheral circulation. It has been suggested by pre-clinical studies that it would take more than 10 L of blood to produce enough EPCs for angiogenesis in a patient. In order to overcome this problem, cell proliferation becomes important for amplifying the pool of EPCs in vitro. 23 Tepper et al. 24 isolated EPCs from type 2 diabetics and demonstrated a 48% decrease in proliferation relative to age-matched control subjects. Chen et al. 25 isolated EPCs from peripheral blood of healthy young human volunteers and incubated them with high glucose in vitro. They found that high glucose–inhibited EPC proliferation and the capacity of colony formation in a dose- and time-dependent manner, and this inhibition became maximal at 25-mM glucose incubation for a long term (⩾ 4 days). In addition, the high glucose–induced EPC proliferation inhibition was accompanied by a decreased level of endothelial nitric oxide synthase (eNOS), FoxO1, Akt phosphorylation and NO release and could be reversed by co-incubation with NO donor sodium nitroprusside or p38 mitogen-activated protein kinase inhibitor. Interestingly, antioxidants could not reverse high glucose–induced EPC proliferation inhibition, some of them like pyrrolidine dithiocarbamate, diphenyleneiodonium, apocynin and rotenone even accelerated this kind of inhibition, which suggested that high glucose–reduced EPC proliferation was mediated by a NO-related mechanism not oxidative stress-related mechanisms.
After 1 year, Zhang et al. reported the same time-dependent responses of EPCs proliferation to high glucose. In the early stage of incubation (3 days) with high glucose, EPC proliferation was promoted and the expression of cdk2 and cyclin E was upregulated, while in the late stage of culture (7 days), the cell proliferation was inhibited, the expressions of cdk2, cyclin E as well as proliferating cell nuclear antigen (PCNA) were decreased. At the same time, the reactive oxygen species (ROS) generation was increased and the expressions of superoxide dismutase (SOD) and glutathione levels decreased in high glucose–cultured EPCs on the seventh day. Moreover, adding radical scavenger to the high glucose medium could partially reverse high glucose–induced EPC proliferation inhibition. All these observations suggest this biphasic response of EPC proliferation may be mediated by the production of ROS. 26
The findings of Zhang et al. seem disagreement with the results obtained by Chen’s group. Different species and EPC isolation procedures between these two groups may be the primary reasons leading to this discrepancy. Moreover, different antioxidant drug treatments may also be important factors as well. Above all, these findings give insights into the complex mechanisms of EPCs for impaired proliferation in long-term exposure to high glucose.
Adhesion and migration
The adhesion of EPCs to both activated ECs and the extracellular matrix is a key step in new blood vessel growth. It has been shown that the adhesion of diabetic EPCs to fibronection, collagen and quiescent ECs was not affected while its adherence to human umbilical vein ECs activated by tumour necrosis factor-α (TNF-α) was decreased. 24 This observation provided insight into the mechanism of EPC function in vivo, suggesting the recruitment of EPCs to the endothelium during the initial phases of inflammation was impaired in diabetic mellitus.
The migration of EPCs in response to VEGF is important during wound healing and neovascularization. 27 Chen et al. 25 evaluated the effect of high glucose on EPC migration using a modified Boyden chamber assay with VEGF as the chemoattractic factor. They observed VEGF-induced EPC migration was significantly reduced, by 37.0%, in the high glucose–treated group as compared to the control one. This high glucose effect could be ameliorated by NO donor co-incubation, suggesting the possible role of NO-related mechanisms in hyperglycaemia-induced EPC migration inhibition.
Tubulization
EPCs can form tube-like structure on Matrigel and incorporate into the mature ECs participating in the new vessel formation, which is essential for tumour vasculogenesis and wound healing. In diabetic patients, the tubulization of EPCs seems abnormal. In a Matrigel assay, diabetic EPCs were 2.5 times less likely to participate in tubule formation compared with controls. 24 During coculture of EPCs isolated from peripheral mononuclear cells from healthy subjects and HUVECs on Matrigel in high glucose conditions (33 mM), a lower amount of EPCs participated into tube formation when compared with controls (normal glucose condition). 21 To quantify this impaired tube formation ability of high glucose–cultured EPCs, Chen et al. 25 compared the capacity for tube formation of EPCs isolated from healthy human subjects and treated with high glucose for 4 days with the control normal glucose-treated cells and revealed that tube formation of high glucose–treated EPCs was significantly reduced, by approximately 38%.
By establishing a novel quantitative methodology, the in vitro 5-(6)-carboxyfluorescein diacetate succinimidyl ester-labelling vessel formation assay, Jiraritthamrong et al. demonstrated the impaired vessel-forming capacity of EPCs cultured under high glucose condition, which was accompanied by a lower level of angiopoietin 1 gene expression. Furthermore, this impairment of tube forming capacity could be reversed by an addition of recombinant angiopoietin 1 to the high glucose medium, 28 which suggests the possible mechanism in regulating EPC tube formation under high glucose conditions.
Senescence
EPC senescence limits their proliferative potential and contributes to endothelial dysfunction and vascular complications in diabetes. Chen et al. 25 determined the age of EPCs isolated from healthy subjects and demonstrated that incubation of either early (attached) or late (monolayer) EPCs with 25-mM high glucose for 4 days significantly enhanced their senescence. In order to find the underlying pathways involved in high glucose–induced EPC senescence, Yuan et al. 29 evaluated EPC senescence by a senescence-associated β-galactosidase (SA-β-Gal) activity assay. They observed an increased percentage of SA-β-Gal positive cells which presents accelerated senescence of EPCs in type 2 diabetic patients in comparison with those in control group. Moreover, the expression of SIRT1 was found to be decreased concomitantly with a decreased level of dimethylarginine dimethylaminohydrolase (DDAH2) and an increased plasma level of ADMA, a novel risk factor for endothelial dysfunction, in type 2 diabetic patients. Interestingly, this phenomenon could be reversed by adding exogenous resveratrol (a SIRT1 activator) in vitro. All these results supported their hypothesis that high glucose–induced EPC senescence may be regulated by SIRT1-DDAH2/ADMA pathway.
Secretion
EPCs play important roles in vascular repair both by differentiating into ECs and by secreting vasoactive substances such as matrix proteins, growth factors and cytokines to promote angiogenesis and maintain vascular haemostasis. 30 Mature ECs incubated with conditioned medium obtained from early EPC cultures exhibited lower levels of intracellular oxidative stress, elevated antioxidant enzymes (catalase, Cu/ZnSOD and MnSOD) expressions and reduced apoptotic rates, which suggest that the secreted factors by early EPCs could protect themselves and mature ECs against oxidative stress in a paracrine manner. 31 Additionally, the late EPCs, named after their outgrowth potential to differentiation into mature ECs and showing cobblestone appearances, also secret cytokines and matrix metalloproteinases (MMPs) to participate in neovascularization after ischaemia injury. 32 It has been reported by Zhang et al. 33 that high glucose impaired EPC secretion functions by lowering levels of vasodilator NO, tissue plasminogen activator (t-PA), plasminogen activator inhibitor-1 (PAI-1), prostaglandin I2 (PGI2) and VEGF, decreasing the activity of SOD and providing possible mechanisms by which EPCs exhibit impaired vascular repair in diabetic patients.
Mobilization and homing
The mobilization of EPCs from bone marrow (BM) to circulation and their homing to ischaemia sites are essential to vasculogenesis during postnatal neovascularization and wound healing. 7 The EPC mobilization process starts with peripheral hypoxia–induced tissue release of VEGF-A, which activates BM stromal NOS and increases NO levels. 36 At the tissue level, ischaemia-induced upregulation of stromal cell-derived factor-1α (SDF-1α) stimulates circulating EPC recruitment to the site. 37 It has been shown in non-diabetic wild-type mice that hyperbaric oxygen (HBO) therapy increases BM NO in vivo via a NOS-mediated mechanism thereby increasing release of EPC into circulation. 38 Later, the impaired phosphorylation of BM eNOS in diabetic mice was revealed by Gallagher et al., 39 resulting in approximately 50% deduction in circulating EPCs. Interestingly, HBO therapy showed a 1200-fold rise in BM NO levels in non-diabetic mice, while the number decreased to 800-fold increase in diabetic mice. The number of circulating EPCs increased 18 h after this kind of HBO therapy in diabetic mice. Additionally, the expression of SDF-1α in cutaneous wounds and EPC homing were also diminished in diabetic mice. Fortunately, administration of extra SDF-1α could reverse the EPC recruitment and, with hyperoxia, synergistically enhanced EPC mobilization, homing and final wound healing. These results provide new insight into the enhancement of diabetic wound healing.
Poor EPC mobilization in response to chemotherapy supplemented with human recombinant granulocyte colony stimulating factor (hrG-CSF) was statistically associated with diabetes, 40 and it was first assayed by Fadini et al. 41 They quantified CPC counts and evaluated their proangiogenic capacity using the Matrigel plug assay. The impaired mobilization of CPCs in response to hrG-CSF was observed, which may be attributed to maladaptive response of the chemokine (C-X-C motif) ligand (CXCL12)-cleaving enzyme dipeptidyl peptidase-4 (DPP-4). Fortunately, this impaired CPC mobilization in diabetes was not observed in diabetic patients who received hrG-CSF plus plerixafor, 42 a C-X-C receptor type 4 (CXCR4) antagonist, which was promising in cardiac and peripheral ischaemic diseases treatment.
Possible molecular mechanisms underlying the reduced number and impaired function of EPCs exposed to high glucose conditions
ROS
The mechanisms of DM-induced EPC deficit are still unclear (Figure 2). However, increased production of ROS by the mitochondria seems to play a crucial role. It has been reported that chronic high glucose exposure of human ECs could elevate the mitochondrial ROS level 43 and decrease the expression and activity of eNOS, 44 which is believed to associate with the diabetic vascular complications. Interestingly, high glucose–induced eNOS expression and activity reduction were mediated by mitochondrial ROS and regulated by oxidative stress transcription factor, activator protein-1 (AP-1). 45 For EPCs, they expressed higher intrinsic antioxidative enzymes, such as manganese superoxide dismutase (MnSOD) than the mature ECs, to protect against oxidative stress.46,47 However, reduced number, differentiation and function of EPCs accompanied by impaired angiogenesis are still happening in diabetic patients or high glucose incubated EPCs. This may be associated with the oxidative stress adaptor protein P66ShcA, one isoform of ShcA with an additional domain at its N-terminus to regulate ROS metabolism and apoptosis,48,49 which is upregulated in high glucose–cultured mouse BM–derived EPCs. 50 Moreover, knockout of P66ShcA could rescue high glucose–induced mouse BM–derived EPCs defect, 50 indicating high glucose impaired EPC number and function with an oxidative-stress-dependent mechanism, and P66ShcA as a potential therapeutic target in diabetic vasculopathy.
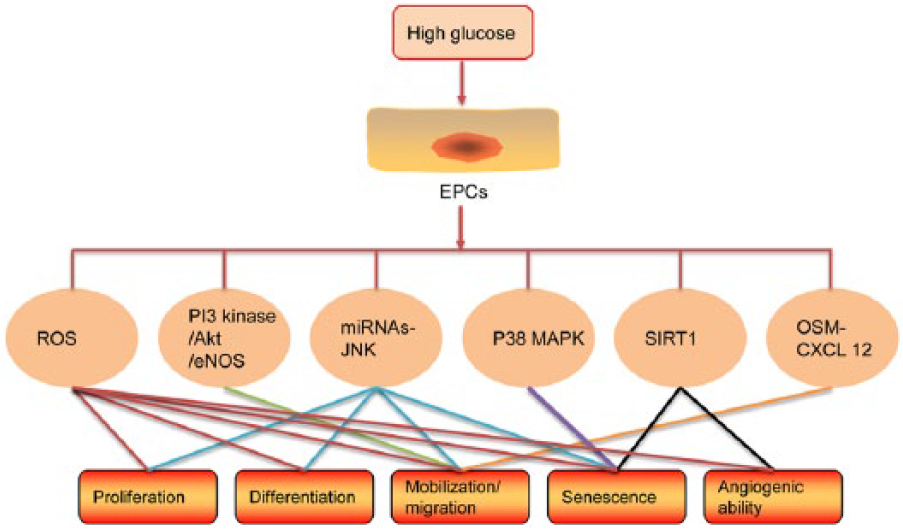
Possible molecular pathways regulating EPCs proliferation, differentiation, mobilization, migration, senescence and angiogenic ability under the condition of high glucose.
PI3 kinase/Akt/eNOS pathway (mobilization/migration)
PI3 kinase/Akt/eNOS pathway is one of the typical signal transduction pathways in regulating EPC mobilization from BM to sites of ischaemia and hypoxia, which could be triggered by SDF-1α and stimulated by the CXCR4.37,51 The importance of NO production and NOS activity in EPC mobilization has been shown by many. Thum et al. 52 reported uncoupling of the eNOS in diabetic BM, high glucose–treated EPCs or EPC from diabetic patients resulted in superoxide anion (O2−) formation, which subsequently reduced EPC levels and impaired their function. Chen et al. 25 demonstrated high glucose dose dependently reduced the number and proliferation of early and late EPCs, enhanced EPC senescence and impaired the migration of late EPCs, which was regulated by a NO-related mechanism. Segal et al. 53 showed that incubating EPCs isolated from diabetic patients with NO donor could correct their migration defect. Recently, in type 2 diabetic patients with coronary artery disease (CAD), hyperglycaemia and hyperlipidaemia impaired EPC number, migration, NO bioavailability and NOS activity, which was accompanied by the downregulation of the expressions of CXCR4 and members of PI3 kinase/Akt/eNOS signal cascades. 54 These results provided the evidence for the possible underlying mechanism for hyperglycaemia impaired EPC migration in type 2 diabetic mellitus with CAD.
MicroRNA–c-Jun NH2-terminal kinase pathway
microRNAs (miRNAs), the small non-coding RNAs, could bind to the 3′-untranslated region (UTR) of target cellular mRNA, resulting in target mRNA degradation or translation inhibition. 55 In this manner, miRNA regulates cell growth, differentiation and apoptosis. Many miRNAs have been reported to be involved in diabetic complications associated with EPC dysfunction. As an example, miRNA 107 has been shown to partly inhibit EPC differentiation via hypoxia-inducible factor-1β (HIF-1β). 56 MiRNA-34a overexpression could increase EPC senescence by SIRT1 and upregulating its effector acetylated FoxO1. 57 In addition, the downregulation of miRNA-126 in EPCs isolated from diabetic patients mediated the impairment of EPC proliferation, migration and apoptosis via its target Spred-1. 58
As a subgroup of the mitogen-activated protein kinases (MAPK) signal pathway, c-Jun NH2-terminal kinase (JNK) pathway is usually activated by stimulating with environmental stresses, such as oxidative stress and osmotic pressure. 59 It plays critical roles in regulating various cell processes including cell proliferation, development, apoptosis, differentiation and inflammatory response. 60 Furthermore, activation of JNK has been shown to be involved in the pathogenesis of diabetes, atherosclerosis and insulin resistance. 61 A most recent study from Ye et al. 62 indicated that in diabetic EPCs, high glucose downregulated the expression of miRNA-130a, which subsequently activated the JNK pathway by targeting the JNK upstream specific activator, MAP3K12. This observation advances our understanding of the role of miRNAs in EPC dysfunction in diabetic mellitus.
p38 MAPK (senescence)
MAPKs, the family of serine/threonine kinases that can be divided into three subgroups including extracellular signal-regulated kinase (ERK), JNK and p38 MAPK, regulate cellular proliferation and differentiation induced by a variety of cellular stresses. It has been shown that high glucose (12.5 mM) accelerated the senescence of cultured EPCs, which was associated with an increase in the phosphorylation level of p38 MAPK in EPCs exposed to high glucose and could be significantly inhibited by an extra addition of p38 MAPK inhibitor, SB203580, suggesting the possible intracellular signal transduction involved in p38 MAPK. 63
SIRT1
SIRT1 NAD+-dependent protein deacetylase, the homologue of silent information regulator-2 (Sir2), regulates cell cycle, senescence, apoptosis, metabolic pathways by interacting with FoxO and other transcription molecules.64,65 In vascular system, SIRT1 plays critical protective roles in stress-induced senescence, 66 promoting endothelium-dependent vasodilation 67 and controlling angiogenic activity of ECs 68 via deacetylation of eNOS, FoxO1 and other molecules. Disruption of SIRT1 expression has been shown to result in defective blood vessel formation in ischaemia-induced neovascularization. 68 The relative contribution of SIRT1 in high glucose–induced EPC number and function impairment was recently evaluated. Treatment of EPCs with high glucose for 3 days reduced EPC number accompanied by changes in the expressions of cell metabolism, cell cycle and oxidative related genes, which was associated with reduced SIRT1 expression levels and enzyme activity and increased acetyl-FoxO1 levels. 69 In addition, downregulation of SIRT1 expression was reported to be associated with impaired angiogenic function and accelerated senescence in vitro. 70 These observations verified the essential modulator role of SIRT1 in hyperglycaemia-induced EPCs dysfunction in diabetes.
Oncostatin M–CXCL12 pathway
As mentioned above, diabetes affects BM structure and impairs stem cell (SC) mobilization, 71 paving the way to multiorgan complications. Oncostatin M (OSM) is a soluble mediator, secreted by diabetic BM macrophages (M1 macrophages in human or CD169+ BM macrophages in mice), which induces CXCL12 expression by mesenchymal stem/stromal cells via a MAPK kinase-p38-signal transducer and activator of a transcription-3-dependent pathway. 72 The CXCL12 induction finally prevents SC mobilization. 40 It has been demonstrated that OSM neutralization in diabetic mice prevented CXCL12 induction, improved G-CSF and ischaemia-induced mobilization and promoted SC homing and vascular repair. In diabetic patients, the OSM plasma levels correlated with the BM-to-PB SC ratio. 72 All these studies support the conclusion that OSM neutralization will be a candidate therapy to restore SC mobilization and vascular repair in diabetes.
Agents alleviate dysfunction of EPCs induced by high glucose
Vaspin
Vaspin (visceral adipose tissue–derived serpin) is an adipocytokine, which is a member of serine protease inhibitor family (Table 2). It is first isolated from visceral white adipose tissues (WATs) of a rat model of abdominal obesity with type 2 diabetes, namely, Otsuka Long–Evans Tokushima fatty (OLETF) rat. Vaspin comprised 392–395 amino acids, with about 40% α1-antitrypsin homology. 73 As an anti-diabetic factor, it inhibits expression of proinflammatory adipocytokines, for example, leptin, resistin, TNF-α, glucose transporter-4 and adiponectin in WATs of OLETF rat. In vascular cells, vaspin shows antiatherogenic properties relying on its preventive effects on free fatty acid (FFA)–induced endothelial apoptosis through Akt-mediated pathway and positive effects on NO bioavailability by reducing ADMA level through STAT3-mediated DDAH2 upregulation mechanism.74,75 Recently, Sun et al. 76 reported that vaspin can alleviate high glucose–decreased EPC proliferation and migration. In addition, it increased the expression and activation of eNOS via a PI3K/Akt/eNOS pathway, which suggests the promising effect of vaspin on improving the dysfunction of EPCs in diabetic mellitus patients for preventing the occurrence of vascular complication.
Agents that alleviate dysfunction of EPCs exposed to high glucose.
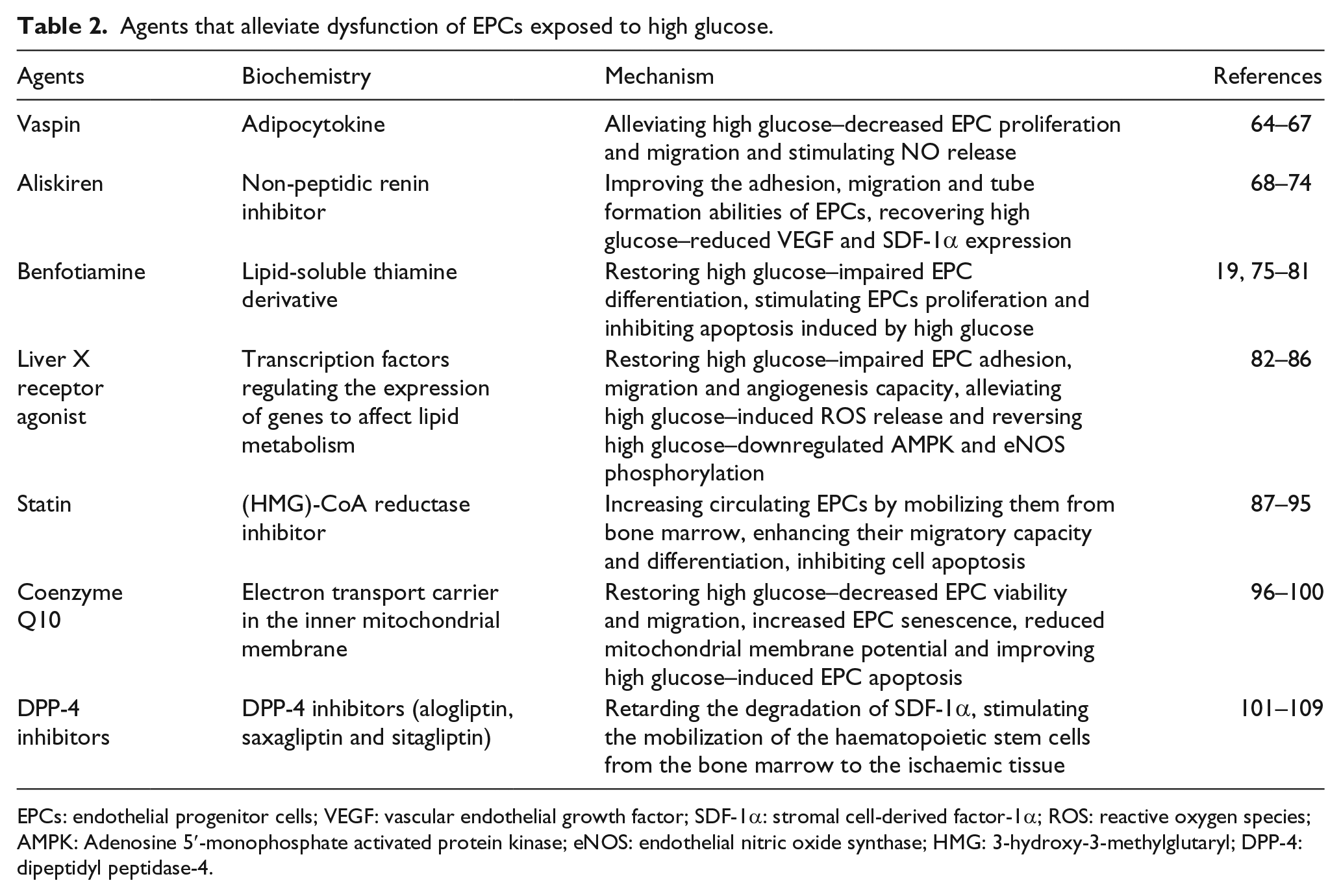
EPCs: endothelial progenitor cells; VEGF: vascular endothelial growth factor; SDF-1α: stromal cell-derived factor-1α; ROS: reactive oxygen species; AMPK: Adenosine 5′-monophosphate activated protein kinase; eNOS: endothelial nitric oxide synthase; HMG: 3-hydroxy-3-methylglutaryl; DPP-4: dipeptidyl peptidase-4.
Aliskiren
Aliskiren is an orally active non-peptidic renin inhibitor, with its 50% inhibitory concentration [IC50] in a low nanomolar range, which can directly reduce angiotension (Ang) II levels. 77 Like all renin-antiotensin system (RAS) blockers, renin inhibition with aliskiren increases a greater rise in the concentration of renin. It has been investigated by Batenburg et al. 78 that this kind of renin surge after aliskiren treatment may be attributed to the two phenomena: first, aliskiren binding converted the prorenin conformation from ‘closed’ to ‘open’, thus allowing its recognition by the active site-specific antibodies in renin assay. This recognition of prorenin as renin may particularly contribute to the overestimation of renin. 79 Second, aliskiren treatment decreases renin/prorenin clearance by increasing the intracellular half life of prorenin two to three times. Aliskiren was shown to reduce prorenin receptor (PRR) expression in diabetic triglycerides (TG) (mRen-2)27 rats and have the binding ability to the active site of prorenin, 80 blocking prorenin-induced angiotensin generation, which suggests an antihypertensive and renoprotective effects of aliskiren in experimental diabetic animals.80,81 In addition, aliskiren exerts synergistic protective effects against ventricle remodelling in combination with valsartan after myocardial infarction (MI) in mice. 82 However, its safety and efficacy in clinical post–MI patients need further assessment.
Very recently, Chang et al. 83 showed that 0.1 or 10-µM aliskiren dose dependently improved the adhesion, migration and tube formation abilities of EPCs isolated from type 2 diabetic patients or healthy volunteers treated with 25-mM high glucose for 4 days. Moreover,10-µM aliskiren treatment recovered high glucose–reduced VEGF and SDF-1α expression, which could be impaired when EPCs were transfected with (P)RR siRNA. These results demonstrated that aliskiren could directly improve the function of EPCs from patients with type 2 diabetes via a (P)RR-VEGF/SDF-1α pathway. Further in vivo and in vitro studies may be performed to clarify the interaction between aliskiren and (P)RR before it could be used in clinic.
Benfotiamine
Benfotiamine, a lipid-soluble thiamine derivative, has the ability to convert glycolytic metabolites glyceraldehyde-3-phosphate and fructose-6-phosphate into pentose-5-phosphates and other sugars by activating pentose phosphate pathway enzyme transketolase, thus blocking three major pathways [the hexosamine pathway, the advanced glycation end (AGE) product formation pathway and the diacylglycerol (DAG)–protein kinase C (PKC) pathway] implicated in the pathogenesis of high glucose–induced vascular damage. 84 This ability of benfotiamine might be clinically useful in preventing the development and progression of diabetic complications. For example, incipient nephropathy, characterized by persistent microalbuminuria and hyperfiltration, is a common complication of diabetes. 85 High plasma glucose concentration in both type 1 and type 2 diabetes is a risk factor for the development of incipient nephropathy by enhancing the cytosolic glucose concentration in renal ECs and accumulating triosephosphates to trigger biochemical dysfuntion, for example, activation of protein kinase Cβ, mitochondrial dysfunction and oxidative stress or accumulation of AGEs. 86 It has been reported that high-dose benfotiamine therapy could convert triosephosphates to ribose-5-phosphate, thus inhibits the occurrence of microalbuminuria, and suppress hyperfiltration, showing potential novel strategy for the prevention of incipient diabetic nephropathy. 110 In addition, benfotiamine is effective in curing diabetic cardiomyopathy, another common diabetic complication with reduced cardiac compliance, 87 by alleviating oxidative stress without affecting AGE or protein carbonyl formation. 88
The involvement of EPCs in diabetic complications has been highlighted in recent years. Benfotiamine was shown to be effective in reversing hyperglycaemia-induced EPC dysfunction by Marchetti et al. 21 When human EPCs isolated from peripheral blood mononuclear cells of healthy subjects were cultured in high glucose (33 mM) medium, their abilities to differentiate into mature ECs were impaired significantly. However, the high glucose medium supplemented with benfotiamine can restore the glucotoxicity via the modulation of Akt/FoxO1 activity.
Moreover, benfotiamine could stimulate the proliferation of human EPCs under high or physiological glucose conditions, inhibit apoptosis induced by high glucose and increase the number of circulating EPCs in diabetic mice submitted to limb ischaemia, being of benefit in reparative neovascularization in type 1 diabetes model of hindlimb ischaemia. 89 In addition, benfotiamine supplementation was reported to prevent diabetes–induced BM microenvironment alteration including microangiopathy, hypoperfusion and lineage (-) c-Kit (+) Sca-1 (+) cell depletion through boosting the antioxidative pentose phosphate pathway. 90
Liver X receptor agonist
Liver X receptors (LXRs) are members of the nuclear receptors that represent a large super family of transcription factors regulating the expression of genes to affect lipid metabolism. 91 LXRs control the transcription of a large number of genes participated in cholesterol metabolism, transport and elimination. 92 As a synthetic LXR agonist, TO901317 (TO) administrated to low-density lipoprotein receptor (LDLR)-/- mice could reduce the atherosclerotic lesions without affecting plasma total cholesterol levels, 93 and these anti-atherosclerotic effects seem different in two arterial regions: the innominate artery and aortic sinus. 94 Recently, Li et al. 95 explored the effect of TO on EPC biology exposed to high glucose (30 mM) condition and revealed that 10 µM pre-treatment of TO with EPCs restored high glucose impaired their adhesion, migration and angiogenesis capacity, alleviated high glucose–induced ROS release and reversed high glucose–downregulated adenosine 5′-monophosphate activated protein kinase (AMPK) and eNOS phosphorylation.
Statin
Statins, 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitor, have been developed as blood cholesterol lowering drugs and performed well in reducing death and ischaemic events from CAD. 96 Moreover, statins possess favourable effects independent of lipid reduction including reducing vascular inflammation, 97 decreasing platelet aggregation and thrombus deposition 98 and increasing endothelium-derived nitric oxide production. 99 Statins have been reported to promote angiogenesis in ischaemic limbs of normocholesterolemic rabbits in an Akt-dependent manner earlier in this century by Kureishi et al. 100 Thereafter, two independent groups have found evidence in both animals 111 and human subjects112,113 that statin treatment increased circulating EPCs by mobilizing them from BM, enhancing their migratory capacity and differentiation, inhibiting cell apoptosis. In line with these studies, Walter et al. revealed that statin therapy might also accelerate reendothelialization of the balloon-injured rat arterial segments and reduce neointimal thickening by increasing the number of EPCs, enhancing their migration to the sites of vascular injury and inducing adhesive molecules expression. 114
More recently, the effect of statin therapy on the EPCs mobilization from the BM into the circulating blood in a diabetic porcine model was evaluated. 115 Results showed that 1 month after diabetic mellitus induction, both BM and circulating EPCs were significantly reduced, compared to the baseline. At this moment, diabetic pigs were given statin therapy. After 3 months of statin administration, the circulating EPCs were increased, and flow-induced vascular dilation was also enhanced approximately 6% relative to the control pigs. These results suggest diabetic mellitus may lower both BM and circulating EPCs which may be partially ameliorated by statin treatment. The beneficial cholesterol independent effects of statins on EPCs in the state of diabetic mellitus provide new insight in the EPCs dysfunction restoration to protect from diabetic complications.
Coenzyme Q10
Coenzyme Q10 was discovered in 1957 by Crane et al. 116 as an important electron transport carrier from complex I or complex II to complex III in the inner mitochondrial membrane to maintain mitochondrial energy homeostasis. 117 In addition, CoQ10 is involved in a number of aspects of cellular metabolism and has exhibited several new functions including as an endogenous lipid-soluble antioxidant, regulating mitochondrial permeability transition pores, activating mitochondrial uncoupling proteins and modulating the amount of β2-integrins on the surface of blood monocytes. 117 In clinic, CoQ10 shows promising effect when given alone or in addition to standard therapies in hypertension, heart failure, excise-related damage, Parkinson’s disease, asthenozoospermia and preeclampsia in pregnancy. 118 Moreover, Watts et al. 119 reported that CoQ10 supplementation improves endothelial function in patients with type 2 diabetes by improving vascular oxidative stress and increasing nitric oxide release or activation. Most recently, CoQ10 was assessed efficient in restoring high glucose–induced EPC dysfunction in diabetic patients. 120 The following results were obtained: first, high glucose (25 mM) decreased EPCs viability and migration by 25% and 30%, respectively, and increased EPC senescence by 28%, which could all be attenuated by a 24-h-CoQ10 incubation. Second, high glucose–reduced mitochondrial membrane potential of EPCs was reversed by CoQ10 treatment. Third, CoQ10 (10 and 20 µM) therapy inhibited high glucose–induced caspase 3 activation and Bcl-2 downregulation, improving high glucose–induced EPC apoptosis. Finally, CoQ10 improved EPC function and attenuated cell apoptosis via AMPK, NO and HO-1 pathways. In animals, blood flow in ischaemia hindlimbs mice could be partially restored by transplantation with CoQ10-treated EPCs cultured under high glucose conditions. These data substantiated the benefit of CoQ10 on the treatment of EPC dysfunction in diabetic mellitus and its related cardiovascular diseases.
DPP-4 inhibitors
DPP-4 is the prolyl oligopeptidase family of serine proteases that modulates the structure and function of peptides by specifically cleaving their N-terminal dipeptides with a strong preference for proline > alanine > serine as the penultimate amino acid residue. 101 DPP-4 cleaves glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), thus affecting glucose metabolism. SDF-1α is a ligand for G-protein–coupled CXCR4, which triggers the downstream G-protein signalling upon binding and inducing conformational changes in the CXCR4 transmembrane region. 102 Acting as an essential SC homing factor, SDF-1α stimulates the mobilization of the haematopoietic SCs from the BM to the ischaemic tissue, and it is a physiological substrate of DPP-4. Therefore, DPP-4 inhibition is expected to increase SDF-1α bioavailability and activity, with the eventual stimulation of EPCs. 103 The first study provided the rationale to use DPP-4 as an EPC stimulator for vascular repair was performed in mice by Zaruba et al. 104 Then, Fadini et al. 105 demonstrated in a small controlled trial that the 4-week course of oral therapy with DPP-4 inhibitor sitagliptin increases the circulating EPCs in type 2 diabetic patients with concomitant high levels of SDF-1α. Moreover, these effects were not associated with the changes in nitrite/nitrate levels or the reduction in plasma glucose, suggesting they are GLP-1 independent. 105 Most recently, Connelly et al. 106 compared the effects of saxagliptin (DPP-4 inhibitor) with those of liraglutide (GLP-1 receptor agonist) and SDF-1α receptor (CXCR4) antagonist plerixafor on survival following experimentally induced MI in diabetic rats. Mortality was reduced and cardiac function was improved in saxagliptin-treated rats, while antagonism of CXCR4 prevented the improvement in cardiac function in saxagliptin-treated rats and was associated with increased mortality. These results indicated that saxagliptin-mediated DPP-4 inhibition via a non-GLP-1-dependent mechanism, probably through SDF-1α potentiation, improved cardiac function after MI independent of glucose lowering. Up to now, several DPP-4 inhibitors are available such as alogliptin, 107 saxagliptin 108 and sitagliptin. 109 They are known to be non-invasive to retard the degradation of SDF-1α and could be the ideal choice for diabetic cardiovascular complications therapy. Further studies are necessary to elucidate the interaction between DPP-4 and SDF-1α and the underlying mechanism.
Conclusion and future perspectives
Although abundant evidence has shown the reduced number and impaired function of EPCs in diabetic patients, little attention has been paid to the individual high glucose effect on the EPCs physiology and its underlying mechanisms. Especially, the EPCs are surrounded by the circulating blood flow and exposed to flow-associated haemodynamic forces. It has been noticed that the exposure of EPCs to well-controlled shear stress in vitro could affect their bioactivities in terms of proliferation, anti-apoptosis, migration, production of bioactive substances, anti-thrombosis, tube formation and endothelial marker expression. 121 Therefore, considering shear effects seems necessary in evaluating EPC behaviour exposed to high glucose condition. In fact, it has been reported that high glucose can disrupt some surface mechanosensors of ECs, which may in turn affect their behaviour in response to shear force. For example, the degrading of glycocalyx (a surface layer comprised proteoglycans and glycoproteins) by high glucose attenuates shear-induced endothelial hydraulic conductivity alteration.122,123 Similarly, the EPC glycocalyx was shown to be important in regulating EPC homing, proliferation, recruiting and activating at sites of vascular damage.123,124 The future study may concern more about high glucose–induced EPC glycocalyx impairment and its subsequent effect on mechanotransduction of shear stress in unveiling the underlying mechanisms of high glucose–induced EPC dysfunction.
For the EPC assessment, standard cell isolation and culture protocol is necessary to comprise the different results obtained by different groups, and the phenotype of EPCs (early versus late) seems more important than the expression of surface markers in enumeration of EPCs. 15 Moreover, as ageing possesses one of the largest factors for the development of cardiovascular disease, growing evidence has been provided that ageing affects the number and function of EPCs including the migration, homing and engraftment capacity;123,125–127 future studies should consider the age effects in evaluating EPC behaviour in response to therapeutic approaches in diabetic patients.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos 31500763, 11572028 and 11421202) and the 111 Project (B13003).