
Abstract
BK channels are major ionic determinants of vasodilation. BK channel function is impaired in diabetic vessels due to accelerated proteolysis of its beta-1 (BK-β1) subunits in response to increased oxidative stress. The nuclear factor E2-related factor-2 (Nrf2) signalling pathway has emerged as a master regulator of cellular redox status, and we hypothesized that it plays a central role in regulating BK channel function in diabetic vessels. We found that Nrf2 expression was markedly reduced in db/db diabetic mouse aortas, and this was associated with significant downregulation of BK-β1. In addition, the muscle ring finger protein 1 (MuRF1), a known E-3 ligase targeting BK-β1 ubiquitination and proteasomal degradation, was significantly augmented. These findings were reproduced by knockdown of Nrf2 by siRNA in cultured human coronary artery smooth muscle cells. In contrast, adenoviral transfer of Nrf2 gene in these cells downregulated MuRF1 and upregulated BK-β1 expression. Activation of Nrf2 by dimethyl fumarate preserved BK-β1 expression and protected BK channel and vascular function in db/db coronary arteries. These results indicate that expression of BK-β1 is closely regulated by Nrf2 and vascular BK channel function can be restored by Nrf2 activation. Nrf2 should be considered a novel therapeutic target in the treatment of diabetic vasculopathy.
Keywords
Introduction
The large-conductance calcium-activated potassium (BK) channels are expressed in high density in coronary artery smooth muscle cells (SMCs), linking Ca2+ homeostasis with the cell membrane potentials, and is a key ionic determinant in the regulation of vascular tone and myocardial perfusion.1,2 Activation of BK channels by increased intracellular free Ca2+ concentrations gives rise to the spontaneous transient outward currents (STOCs), which hyperpolarize the membrane potentials of vascular SMCs, inactivate the voltage-dependent Ca2+ channels and lead to vasorelaxation.3–5 Functional vascular BK channels are composed of pore-forming α subunits (encoded by the KCNMA1 gene) and accessory β1 subunits (encoded by the KCNMB1 gene) in 4:4 stoichiometry.1,6 The BK-β1 is a key modulator of BK channel electrophysiology, enhancing the BK-α sensitivity to Ca2+ and voltage, allowing channel activation in the physiological ranges of [Ca2+]i and membrane potentials.5,7,8 Regulation of BK channel activity by many biological mediators is mediated through BK-β1 stimulation, demonstrating the importance of BK-β1 in vascular physiology.9,10 A large body of evidence has indicated that vascular BK channel malfunction in diabetes is mainly associated with a significant downregulation of BK-β1 protein expression, resulting in Ca2+ sparks/STOCs uncoupling and loss of Ca2+-mediated channel activation.5,11–14 We have reported that the downregulation of vascular BK-β1 expression in diabetic vessels was attributed to increased protein expression of the muscle ring finger protein 1 (MuRF1, a muscle-specific E3 ligase) that promotes BK-β1 protein degradation via the ubiquitin–proteasome system (UPS) in vascular SMCs. 13 However, the upstream signalling mechanism responsible for the dysregulation of MuRF1 expression in diabetes is unknown.
The Nrf2 signalling has emerged as a master regulator of cellular redox status and detoxification responses. 15 Many antioxidant enzymes, including those of NADPH dehydrogenase quinone 1 (NQO1), glutathione-disulfide reductase (GSR), glutathione translocase (GSTA), thioredoxin (TXN), thioredoxin reductase 1 (TXNRD1), heme oxygenase-1 (HO-1), superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx), are regulated by Nrf2.16–20 Activation of Nrf2 is protective against hyperglycaemia-induced reactive oxygen species (ROS)-mediated apoptosis and cell damage in renal, cardiac and vascular cells. 21 It has been suggested that abnormal regulation of Nrf2 is implicated in the development of a wide range of human diseases, including diabetes. 22 However, the role of Nrf2 signalling in regulating vascular BK channel function in diabetes is unknown. In this study, we hypothesized that Nrf2 plays a central role in the regulation of coronary arterial myocyte BK channel function in diabetes. We found that Nrf2 regulated MuRF1-dependent BK-β1 proteolysis. Molecular and pharmacological activation of Nrf2 preserved BK-β1 expression and protected BK channel-mediated coronary vasodilation in type 2 diabetic db/db mice.
Materials and methods
Type 2 diabetic mice
The type 2 diabetic db/db mice (BKS.Cg-Dock7m+/+Leprdb/J) and age-matched Lean control mice (Dock7m+/Dock7m) were obtained from the Jackson Laboratory. Mice with blood glucose >300 mg/dL were considered diabetic and were used for experiments 10 weeks after developing hyperglycaemia. All protocols were approved by the Institutional Animal Care and Use Committee of the Mayo Clinic, Rochester, MN, USA.
Cell culture, cDNA transfection and adenoviral delivery of Nrf2 and shRNA
HEK293 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco-Thermo Fisher Scientific Inc., Waltham, MA, USA) containing foetal bovine serum (FBS; 10%, v/v), penicillin (100 U/mL) and streptomycin (100 U/mL). Green fluorescent protein (GFP)-tagged Nrf2 cDNA/in pcDNA3 (1 µg) was transfected into HEK293 cells using the Effectene Transfection Reagent kit (Qiagen Co., Valencia, CA, USA). Human coronary arterial SMCs (HCSMCs) were purchased from Lonza Walkersville, Inc. (Walkersville, MD, USA), and were cultured with Clonetics SmBM (Lonza Walkersville, Inc.) containing 5 mM glucose and 17 mM
Reverse transcription polymerase chain reaction and real-time polymerase chain reaction
Total RNA was isolated from SMCs using RNeasy Plus Mini kit (Qiagen Co., Valencia, CA, USA) and was reverse-transcribed to cDNA using SuperScript III First-Strand Synthesis System kit (Invitrogen Thermo Fisher Scientific Inc., Carlsbad, CA, USA). 14 Quantitative gene expression of BK-β1 was determined by reverse transcription polymerase chain reaction (RT-PCR) as the average of triplicates per gene, per cDNA sample.Copy numbers of the target gene were calculated according to 2−ΔCt (where Ct is the cycle threshold and ΔCt = Ct of target gene − Ct of internal control gene, GAPDH). RT-PCR was performed using the iCycler iQ Real Time Detection System (Bio-Rad, Hercules, CA, USA). The reaction underwent a 40-cycle amplification with the following conditions: denaturalization for 15 s at 94°C, annealing for 30 s at 55°C and extension for30 s at 70°C. Oligonucleotide primers were synthesized by IDT Integrated DNA Technologies Inc. (Coralville, IA, USA) with BK-β1 primer sequences: 5′-CACCTGATTGAGACCAACATCAGG-3′ (forward) and 5′-GCTCTGACCTTCTCCACGTC-3′ (reverse); and GAPDH primer sequences: 5′-TGCCAAGGCTGTGGGCAAGG-3′ (forward) and 5′-TGGGCCCTCAGATGCCTGCT-3′ (reverse).
Western blot analysis
Isolated mouse aortas and cultured HCSMCs were homogenized, electrophoresed, transferred to a nitrocellulose membrane and blotted against rabbit anti-Nrf2 (1:200, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; catalogue no. sc-722), anti-NQO1 (1:200, Santa Cruz Biotechnology Inc.; catalogue number sc-16464), anti-BK-β1 (1:200, custom made) 24 and anti-MuRF1 (1:200, Santa Cruz Biotechnology Inc.; catalogue no. sc-32920). Horseradish peroxidase–conjugated secondary antibodies were then added after extensive washing. Signals were developed by Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific Inc.). Blots were also probed with anti-β-actin antibody (1:10,000, Sigma-Aldrich Co. LLC., USA) as loading controls. Optical density of the bands was analysed with Scion Image software (Scion Corp., Frederick, MD, USA). Protein expression was expressed as the relative abundance normalized to β-actin.
Mouse coronary artery SMC isolation and whole-cell BK channel current recording
Single SMCs were enzymatically isolated from the coronary arteries of Lean control and db/db diabetic mice. BK currents from freshly isolated coronary SMCs were recorded using whole-cell patch clamp techniques.11,25 The pipette solution contained (in mM) KCl 140, MgCl2 0.5, Na2ATP 5.0, Na2GTP 0.5, HEPES 10.0, EGTA 1.0, CaCl2 0.465 (~200 nM free Ca2+) at pH 7.38. The bath solution contained (in mM) NaCl 145, KCl 5.6, MgCl2 1.0, CaCl2 1.0, HEPES 10.0, and glucose 5.0 at pH 7.40. BK currents were defined by their sensitivity to 100 nM iberiotoxin (IBTX, a membrane-impermeable BK channel-specific blocker) and were obtained by subtraction of the IBTX-insensitive components from the total K+ currents. The effects of dimethyl fumarate (DMF) on BK current density in coronary SMCs of db/db diabetic mice were evaluated after a 12-h incubation with 10 µM DMF and compared to those of control mice. Experiments were conducted at room temperature (22°C).
Shear stress–mediated coronary vasodilation
Videomicroscopy was employed to measure shear stress (SS)-mediated coronary vasodilation in control and diabetic db/db mice as previously described. 26 Briefly, isolated coronary arteries (1–2 mm in length and 80–130 µm in diameter) from control and db/db mice were isolated by surgical dissection and incubated overnight with DMF (10 µM) or with vehicle [dimethyl sulfoxide (DMSO) at ⩾1000 dilutions]. Vessels were mounted in a temperature-regulated (37°C) chamber filled with Krebs’ solution containing (in mM) NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25 and glucose 11.1 at pH 7.4, secured between two borosilicate glass micropipettes with 10–O ophthalmic sutures, and then placed on the stage of an inverted Olympus CK40 microscopy (Olympus America Inc., Center Valley, PA, USA) equipped with a Olympus OLY-105 CCD camera and a video micrometer (VIA-100, Boeckeler Instruments, Inc., Tucson, AZ, USA). After filling with Krebs’ solution, the intraluminal pressures of mounted coronary arteries were maintained at 80 mmHg. Incremental levels of SS (1, 5, 10, 15, 20 and 25 dynes cm−2) in the vessel were achieved using a microinjection pump and a pressure-servo controller (Living Systems, Burlington, VT, USA) as previously described. 26 Vessels were deemed unacceptable for experiments if they demonstrated leaks, failed to produce a more than 50% constriction to graded doses of endothelin-1 (up to 10 nM) or failed to dilate with a Ca2+-free solution.
Chemicals
Unless otherwise mentioned, all chemicals including DMF were purchased from Sigma-Aldrich Co. LLC. DMF was dissolved in DMSO and diluted with water into 10 mM stocks.
Statistical analysis
Data were expressed as mean ± standard error of mean (SEM). Student’s t test was used to compare data between two groups. A paired t test was used to compare data before and after treatment. One-way analysis of variance (ANOVA) followed by Tukey test analysis was used to compare multiple groups using SigmaStat 3.5 software (Systat Software, Inc., Chicago, IL, USA). Statistically significant difference was defined as p < 0.05.
Results
Metabolic characterization of type 2 diabetic db/db mice
The db/db diabetic mice are an established type 2 diabetic model with leptin receptor deficiency leading to obesity, insulin resistance and diabetes. Typically, the mice begin to have elevated levels of plasma insulin at 10–14 days of age and blood glucose >300 mg/dL at 4–8 weeks of age. At the time of experiments (10 weeks after development of hyperglycaemia >300 mg/dL), the average body weights of Lean control and db/db mice were 26.44 ± 0.72 g and 46.83 ± 1.27 g (n = 20 for both, p < 0.05), respectively. The blood glucose and serum insulin levels were significantly higher in db/db mice compared to age-matched controls: the blood glucose levels were 572.5 ± 13.34 mg/dL in db/db mice versus 148.8 ± 9.11 mg/dL in controls (n = 20 for both, p < 0.05) and insulin levels were 10.50 ± 1.21 ng/mL in db/db mice versus 4.81 ± 1.42 ng/mL in controls (n = 12 for both, p < 0.05).
Nrf2 and BK-β1 are downregulated in diabetic vessels and in HCSMCs cultured in HG
Expression of Nrf2 in diabetic db/db mouse aorta was downregulated by 91% compared to controls (1.0 ± 0.03 in Lean vs 0.09 ± 0.03 in db/db, n = 6, p < 0.05; Figure 1(a)). The NAD(P)H dehydrogenase quinone 1(NQO1) gene, a target of Nrf2 signalling, was reduced by 67.1% (1.0 ± 0.50 in Lean vs 0.33 ± 0.16 in db/db, n = 4, p < 0.05; Figure 1(a)) in diabetic mouse aortas. These changes were associated with a significant downregulation of BK-β1 protein expression by 56.4% (1.0 ± 0.04 in Lean mice, n = 6 vs 0.44 ± 0.09 in db/db mice, n = 6, p < 0.05) and an upregulation of MuRF1 expression by 3.2-fold (1.0 ± 0.11 in Lean vs 3.20 ± 0.21 in db/db, n = 6 for both groups, p < 0.05; Figure 1(a)). These results indicate that Nrf2 signalling is altered in DM and is accompanied by BK-β1 dysregulation. In addition, the BK-β1 mRNA expression was not different between Lean (1.20 ± 0.32 in relative copy numbers, n = 4) and db/db mouse aortas (0.87 ± 0.16, n = 4, p = NS). These results suggest that the downregulation of BK-β1 is due to increase in protein degradation by the ubiquitin–proteasomal system because of enhanced MuRF1 expression.
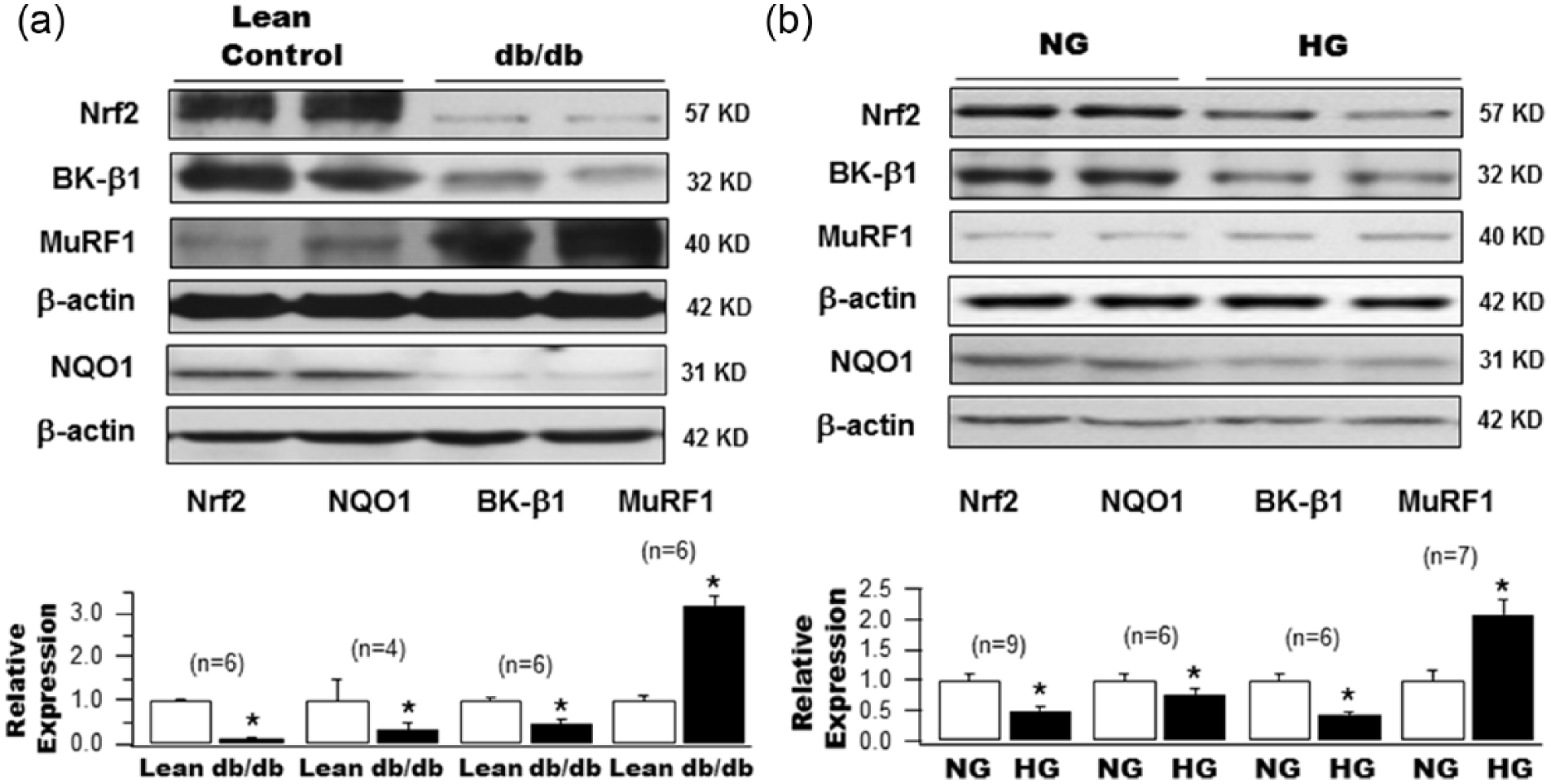
Protein expression of Nrf2, NQO1, BK-β1 and MuRF1 in the aortas of type 2 diabetic db/db mice and in HCSMCs cultured with HG. Western blot analysis of Nrf2, NQO1, BK-β1 and MuRF1 protein expression (a) in the aortas of diabetic db/db mice and age-matched control Lean mice, as well as (b) in HCSMCs 2 weeks after culture with 22 mM glucose (HG) or with 5 mM glucose and 17 mM
A similar profile of changes in the expression of Nrf2, NQO1, BK-β1 and MuRF1 was observed in HCSMCs after a 2-week culture in HG (22 mM), which is the period required for the development of glucotoxicity effects on BK-β1 expression in HCSMCs as previously reported.
14
Compared to NG (5 mM glucose and 17 mM
Molecular knockdown of Nrf2 downregulates BK-β1 and upregulates MuRF1 expression
To further evaluate the regulation of BK-β1 expression by Nrf2, we knocked down Nrf2 expression in human coronary SMCs cultured in NG using Ad-GFP-Nrf2 shRNA. Transduction with adenoviral vector containing scrambled RNAi served as control. After a 48-h transduction, shRNA achieved a 58.9% knockdown of Nrf2 expression (1.0 ± 0.11 with scrambled-RNAi control vs 0.41 ± 0.06 with Nrf2 shRNA, n = 9, p < 0.05) and was accompanied by a 50.9% reduction in NQO1 expression (1.0 ± 0.16 with control vs 0.49 ± 0.07 with Nrf2 shRNA, n = 6, p < 0.05), a 68.9% downregulation of BK-β1 expression (1.0 ± 0.10 with control vs 0.31 ± 0.05 with Nrf2 shRNA, n = 9, p < 0.05) and a 300% upregulation of MuRF1 expression (1.0 ± 0.15 with control vs 3.0 ± 0.51 with Nrf2 shRNA, n = 9, p < 0.05; Figure 2(a)).
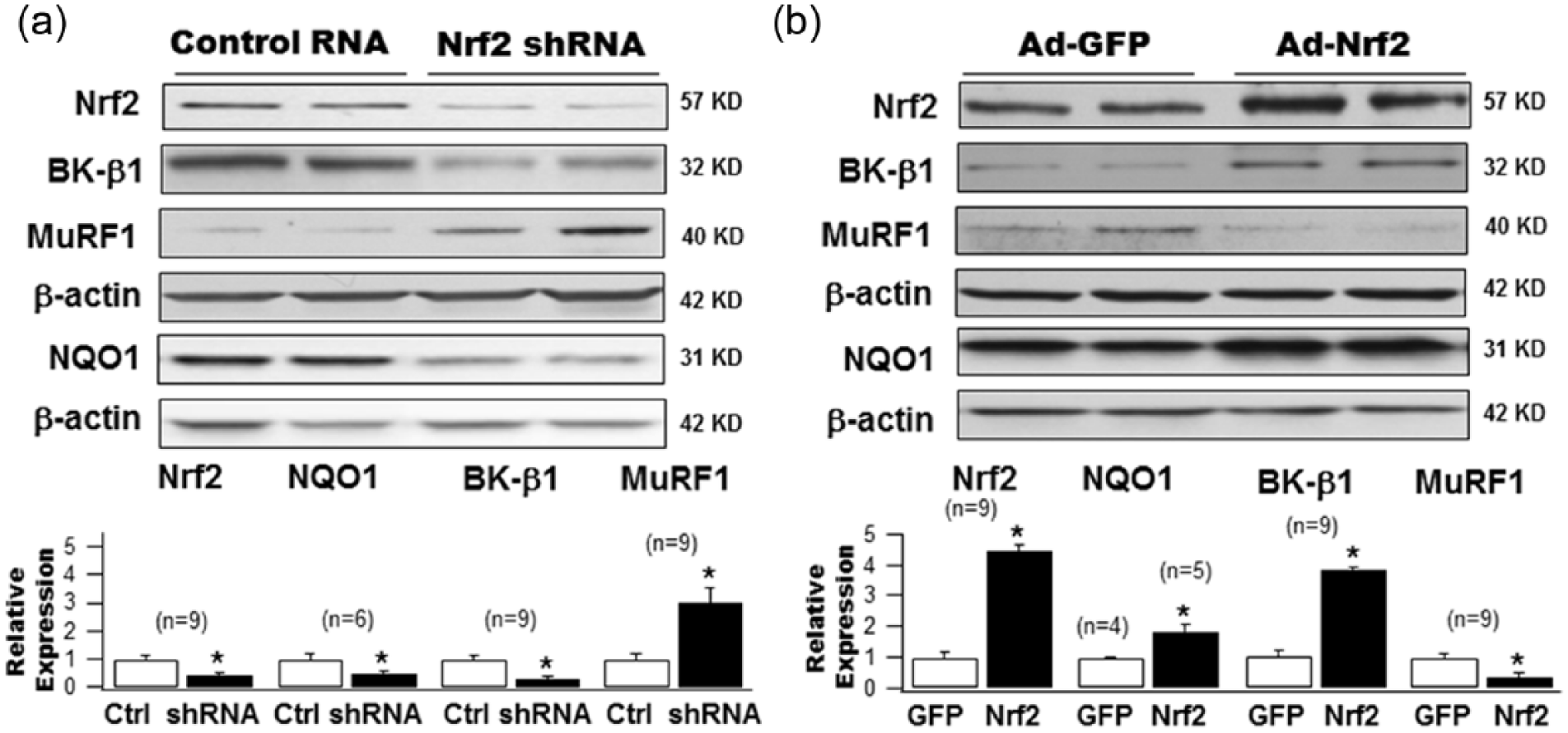
Effects of knockdown and overexpression of Nrf2 on protein expression of BK-β1 NQO1 and MuRF1 in cultured HCSMCs. (a) HG-cultured HCSMCs were transduced with adenovirus carrying the Nrf2 shRNA (Ad-Nrf2 shRNA) or with control adenovirus carrying scrambled RNA (Ad-GFP-U6-Scramble-RNAi) at 50 MOI. A 48-h transduction of Ad-Nrf2 shRNA produced a 58.9% downregulation of Nrf2 expression, a 50.9% reduction in NQO1 expression, a 68.9% reduction in BK-β1 expression but a threefold increase in MuRF1 protein level. (b) A 48-h adenoviral transduction of the Nrf2 gene by adenovirus carrying the GFP-tagged human Nrf2 gene (Ad-Nrf2) markedly augmented Nrf2 expression by 6.8-fold, with a significant increase in NQO1 (1.82-fold) and BK-β1 (2.3-fold) protein expression but a decrease in MuRF1 (45.8%) expression in HG-cultured HCSMCs, compared to cells transduced with Ad-GFP. Group data with statistical significance are shown in the bar graphs.
Molecular overexpression of Nrf2 upregulates BK-β1 and downregulates MuRF1 expression
The effects of Nrf2 overexpression on BK-β1, NQO1 and MuRF1 expression were determined in HCSMCs cultured in HG for 2 weeks. Forty-eight hours after transduction with adenoviral vector containing the Nrf2 gene, Nrf2 protein was overexpressed by 6.8-fold compared to those transduced with empty vectors containing GFP (1.0 ± 0.23 with Ad-GFP vs 6.84 ± 1.07 with Ad-Nrf2, n = 9, p < 0.05; Figure 2(b)). This was accompanied by augmented expression in NQO1 by 1.82-fold (1.0 ± 0.04 with Ad-GFP, n = 4 vs 1.82 ± 0.23 with Ad-Nrf2, n = 5, p < 0.05) and in BK-β1 by 2.3-fold (1.0 ± 0.15 with Ad-GFP vs 2.31 ± 0.40 with Ad-Nrf2, n = 9, p < 0.05). In contrast, MuRF1 was reduced by 45.8% (1.0 ± 0.11 with Ad-GFP vs 0.54 ± 0.11 with Ad-Nrf2, n = 9, p < 0.05; Figure 2(b)). These results confirm that the protein expression of BK-β1 and MuRF1 was tightly regulated by Nrf2 in cultured HCSMCs.
Treatment with DMF upregulates Nrf2, NQO1 and BK-β1 but downregulates MuRF1 expression in HCSMCs
DMF (BG-12, Tecfidera) is a Food and Drug Administration (FDA)-approved Nrf2 activator that has been used successfully in the treatment of multiple sclerosis in the Comparator and an Oral Fumarate in Relapsing-Remitting Multiple Sclerosis (CONFIRM) study.27,28 In HEK293 cells transiently expressing the Nrf2 gene, a 12-h incubation with 10 µM DMF increased the levels of Nrf2 in the nuclear extracts by 4.5-fold, compared to cells treated with vehicle (Figure 3(a)). These results suggest that treatment with DMF promotes the nuclear translocation of Nrf2 and facilitates its function as a transcriptional factor. We further examined the effects of DMF (48-h incubation at 10 µM) on the protein expression of Nrf2, NQO1, BK-β1 and MuRF1 in HCSMCs cultured in HG (Figure 3(b)). DMF treatment enhanced the protein expression of Nrf2 by 1.72-fold (1.0 ± 0.11 with vehicle vs 1.72 ± 0.23 with DMF, n = 3, p < 0.05), that of NQO1 by 2.02-fold (1.0 ± 0.04 with vehicle, n = 3, vs 2.02 ± 0.15 with DMF, n = 5, p < 0.05) and that of BK-β1 by 2.9-fold (1.0 ± 0.13 with vehicle vs 2.90 ± 0.52 with DMF, n = 3, p < 0.05), but that of MuRF1 was reduced by 64.2% (1.0 ± 0.19 with vehicle vs 0.36 ± 0.11 with DMF, n = 3, p < 0.05). These results indicate that pharmacological activation of Nrf2 inhibits MuRF1-mediated BK-β1protein degradation.
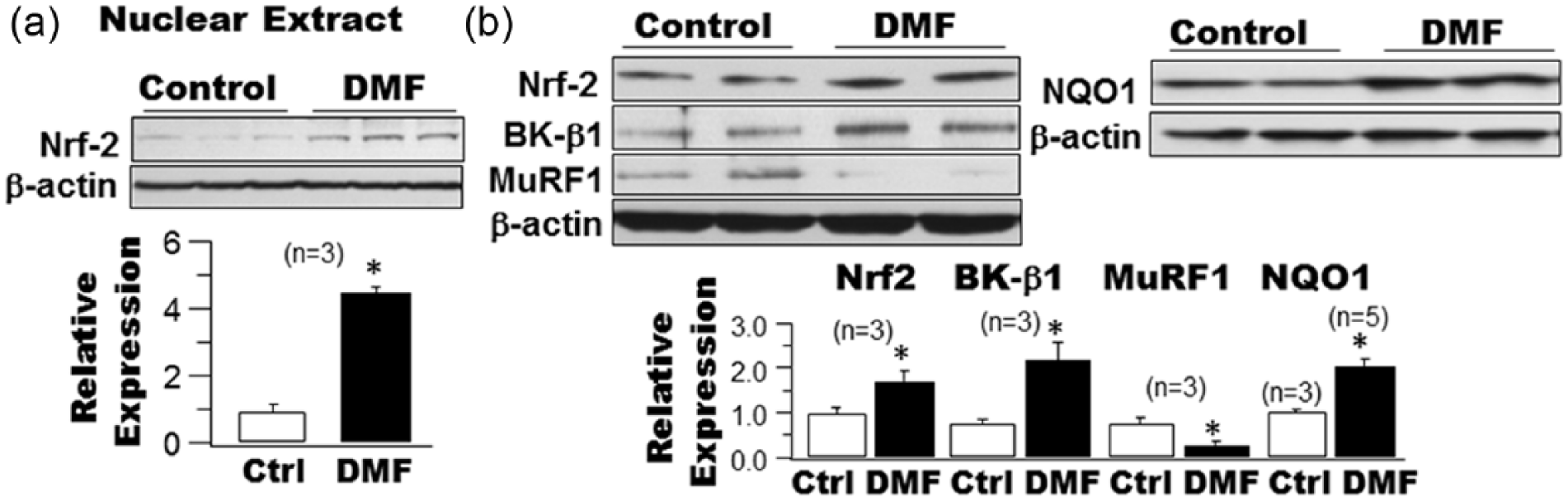
Effects of dimethyl fumarate (DMF) on Nrf2 nuclear translocation in HEK293 cells expressing Nrf2 and on the protein expressions of Nrf2, NQO1, BK-β1 and MuRF1 in cultured HCSMCs.
Activation of Nrf2 by DMF restores BK channel function and vasoreactivity in the coronary arteries of db/db mice
The functional effects of Nrf2 activation by DMF were examined in coronary SMCs freshly isolated from Lean control and db/db diabetic mice. Figure 4 shows representative tracings of IBTX-sensitive K+ current component (BK currents) recorded in coronary SMCs from Lean and db/db diabetic mice 12-h after incubation with 10 µM DMF or with vehicle. Incubation with DMF had no significant effects on BK current densities in coronary SMCs from Lean controls. At a test potential of 130 mV, BK current density was 405.7 ± 22.7 pA/pF (n = 5) with vehicle vs 511.4 ± 200.1 pA/pF with DMF (n = 6, p = NS). In comparison, the BK current density in db/db mice was diminished, exhibiting only 22% of that in controls (89.5 ± 51.1 pA/pF, n = 7, p < 0.05 vs Lean control). However, treatment of the cells from the db/db mice with DMF restored BK current density (425.8 ± 175.9 pA/pS, n = 4, p < 0.05 vs no DMF treatment in db/db), to a normal level similar to that of Lean controls (p = NS vs DMF treatment in Lean controls). The BK current densities of Lean and db/db mice at all testing voltages with and without DMF treatment are shown in the current–voltage (I-V) curves. These results indicate that activation of Nrf2 by DMF has therapeutic implications in preserving coronary BK channel function in db/db diabetic mice.
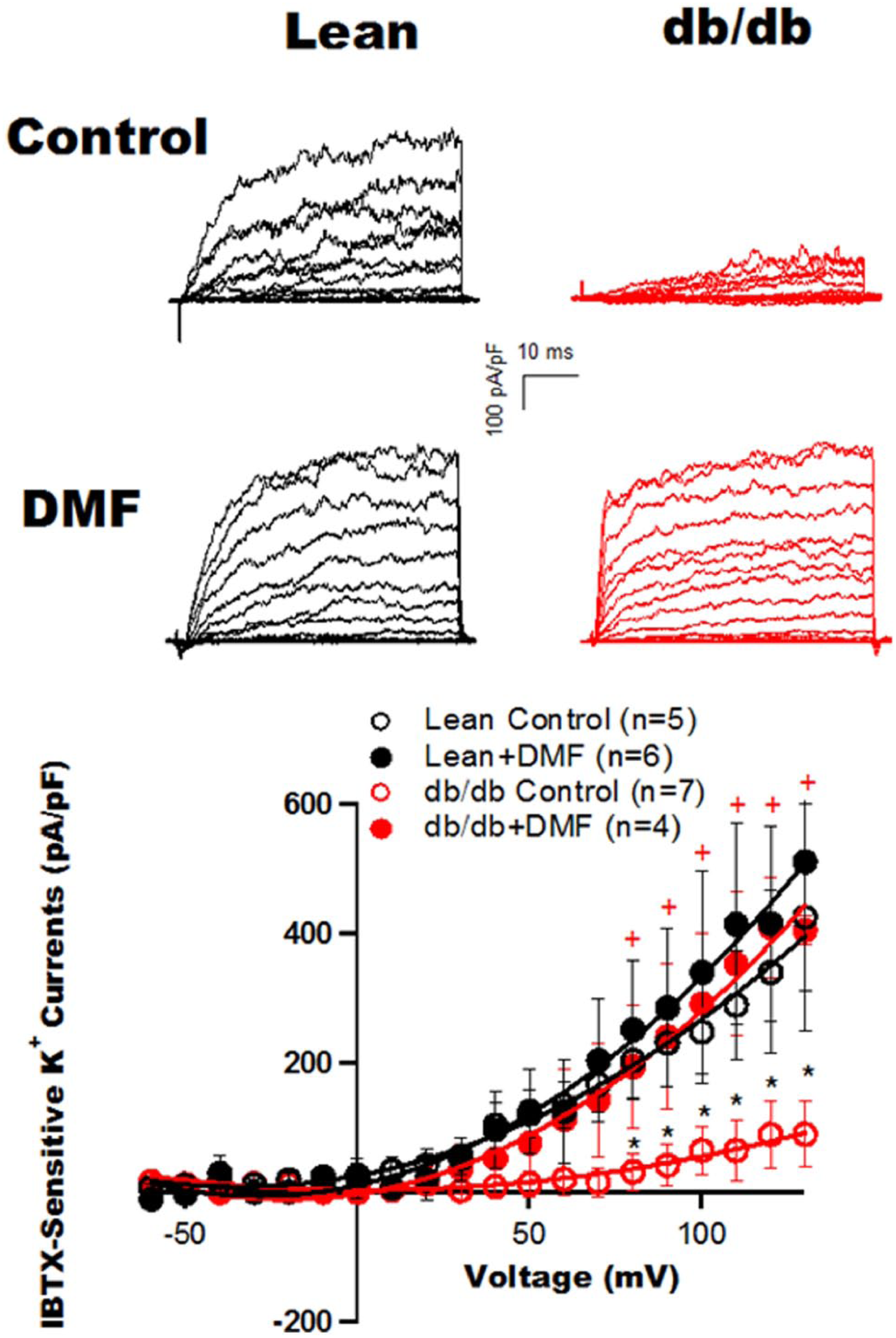
Effects of Nrf2 activation by DMF on BK current densities in coronary SMCs isolated from control and diabetic mice.
Representative tracings of IBTX-sensitive K+ component (BK currents) were recorded in freshly isolated coronary SMCs from type 2 diabetic db/db mice and age-matched Lean control mice with and without DMF treatment (10 µM for 12 h). Recordings were made from a holding potential of −60 mV to test potentials of −60 to 130 mV in 10-mV increments. The BK current density (pA/pF) was markedly reduced in db/db mouse coronary SMCs, compared to that in Lean controls. Incubation with DMF significantly enhanced the BK current density in db/db SMCs, restoring it to a level comparable to that in Lean controls. In contrast, treatment with DMF had no significant effect on cells from Lean controls. The graph shows the BK I-V relationships. *p < 0.05 vs control, +p < 0.05 vs db/db control.
To determine the effects of Nrf2 activation by DMF on vascular function, we examined SS-mediated vasodilation in freshly isolated coronary arteries from control and db/db diabetic mice.26,29 SS-mediated vasodilation was impaired in db/db diabetic coronaries. SS at 25 dynes/cm2 produced 25.2% ± 2.9% vasodilation in db/db mice (n = 3), compared to 76.6% ± 6.6% in Lean mice (n = 3, p < 0.05). Incubation with DMF (10 µM overnight) significantly restored vascular function at all levels of SS. SS at 25 dynes/cm2 produced 53.5% ± 2.1% (n = 4) vasodilation in db/db mouse coronary arteries after treatment with DMF (p < 0.05 vs no treatment). In contrast, treatment with DMF in Lean controls had no effect on SS-mediated vasodilation (Figure 5(a)). In addition, vasodilation by NS-1619 (10−9 to 10−5 M), a BK channel-specific activator, showed significant augmentation after incubation with DMF in db/db vessels (Figure 5(b)). These results are consistent with the patch clamp findings. Hence, activation of Nrf2 by DMF restores coronary vasoreactivity in diabetes and improves diabetic vasculopathy.
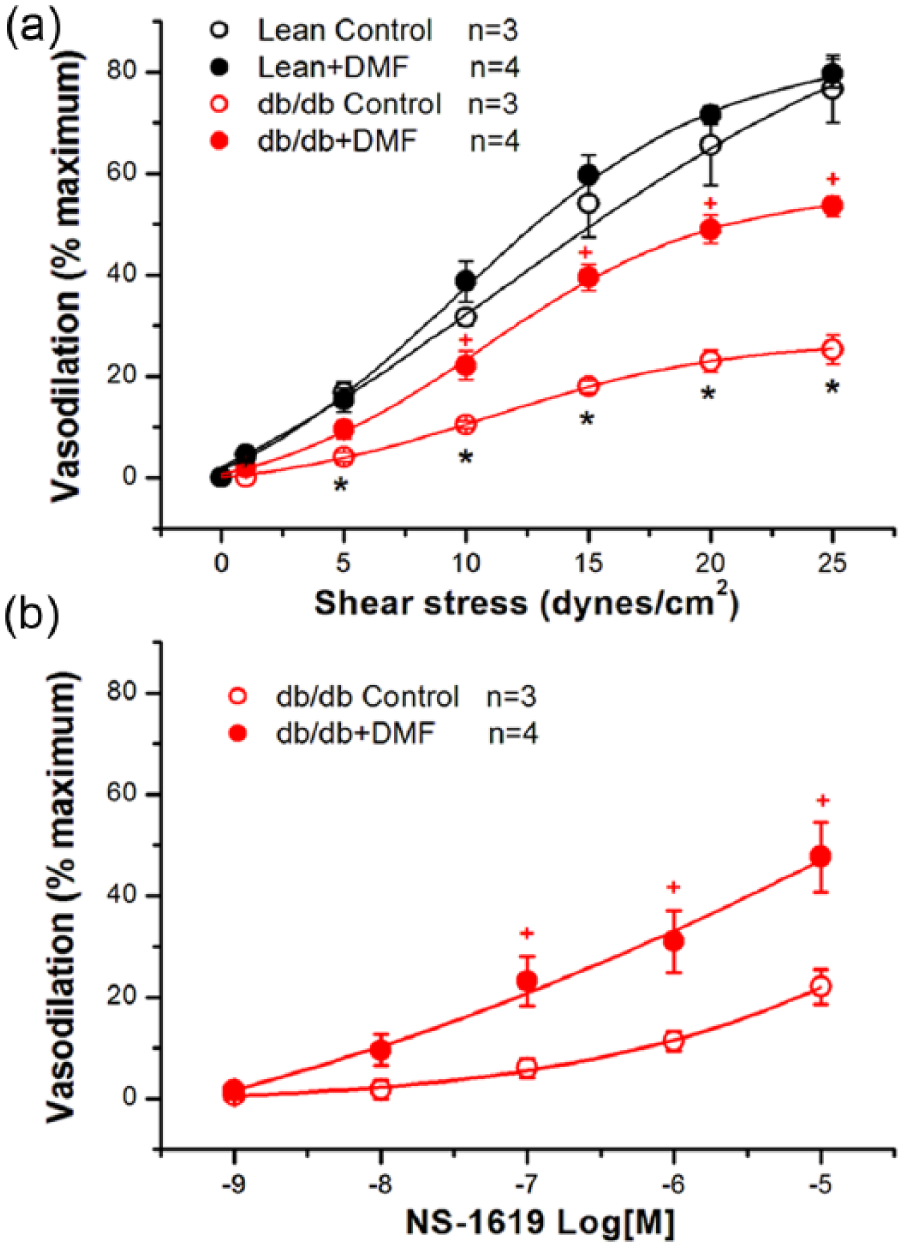
Effects of Nrf2 activation by DMF on shear stress (SS)-mediated and NS-1619-induced coronary vasodilation of diabetic db/db mice. Freshly isolated mouse coronary arteries were incubated with DMF (10 µM) or with vehicle (control) overnight. (a) Vessels were mounted in vessel chambers and subjected to physiological levels of shear stress (0, 1, 5, 10, 15, 20 and 25 dynes/cm2). (b) Mounted vessels were exposed to increasing concentrations of NS-1619 (10−9 to 10−5 M, a BK channel-specific activator). SS-mediated and NS-1619-induced vasodilation is expressed as % maximal vasodilation determined by exposure to zero calcium.
Discussion
There are several important observations in this study. First, expression of Nrf2 is downregulated in diabetic vessels and this diminished the expression of its downstream targets including NQO1 and BK-β1. Second, BK-β1 expression is reduced due to the upregulation in MuRF1 expression, which accelerated the degradation of BK-β1. Third, activation of Nrf2 using molecular and pharmacological approaches upregulates BK-β1 expression through downregulation of MuRF1 expression. Fourth, activation of Nrf2 results in restoration of BK channel function and vascular reactivity in diabetic coronary arteries. These novel findings may have important physiological and clinical implications.
BK channels are critical determinants of vasodilation and are sensitive to the metabolic state and cellular redox environment.11,12,30,31 BK channel function is impaired in both type 1 and type 2 diabetes.11,12,23,25,32–35 The molecular basis of BK channel dysfunction in diabetes is mainly due to the downregulation of BK-β1 expression with loss of BK-β1-mediated activation of the channel.11,25,33 Our results indicate that the reduced BK-β1 level in diabetic vessels is not due to transcriptional failure since mRNA levels were not diminished,13,14 but rather due to accelerated degradation through the UPS with increased levels of BK-β1 ubiquitination in type 2 diabetic db/db mice.
Most (80%–90%) cellular proteins are degraded in a highly specific and selective fashion via the UPS.36,37 Proteins that are targeted for degradation are ubiquitinated through the sequential action of three enzymes: ubiquitin-activation enzyme E1, ubiquitin-conjugating enzyme E2 and ubiquitin-protein ligase E3.36,37 There is one E1 enzyme, more than 25 E2s, and probably more than 1000 E3 ubiquitin-protein ligases. Each E3 recognizes a specific motif on the substrate protein; hence, the specificity of the UPS resides with the E3s, and mutations on specific E3s give rise to specific phenotypes and syndromes associated with abnormal ubiquitin-proteasomal regulation. 38 The polyubiquitinated proteins are then presented to proteasomes for degradation. The UPS is important in regulating many cellular processes, and aberrations of the UPS are implicated in a wide range of disorders including diabetes and cardiovascular diseases such as myocardial ischaemia, ischaemia-reperfusion injuries, 39 atherosclerosis, cardiac atrophy, hypertrophy and hypoxia-induced remodelling of the heart.22,36–38,40–42 Some ion channels are known to be regulated by the UPS.43,44 Recently, we have found that BK-β1 is a target of MuRF1, a muscle-specific E3 ligase, which is significantly upregulated in streptozotocin (STZ)-induced type 1 diabetic vessels and is responsible for accelerating the degradation of BK-β1 in vascular SMCs. 13 Similar findings are observed in type 2 diabetic animals in this study. The MuRF1-mediated downregulation of BK-β1 is dependent on the physical interaction between the N-terminus of BK-β1 and the coil–coil domain of MuRF1. 13 The accelerated BK-β1 degradation in diabetes appears to be facilitated by enhanced ROS and the UPS is responsible for removal of oxidized proteins.45,46 However, the upstream molecular mechanisms resulting in abnormal MuRF1 regulation in diabetes were unknown. In this study, we have identified Nrf2 dysfunction as the underlying defect leading to MuRF1 upregulation and BK-β1 downregulation in type 2 diabetic vessels.
Traditionally, the concept of oxidative stress has been largely ‘superoxide-centric’. 47 In the last decade, our view on oxidative stress in disease mechanisms has significantly broadened with attention to the imbalance between underlying mechanisms of gene expression and regulation that contribute to ROS. At the centre of this new focus is Nrf2 which is considered a master regulator of the antioxidant response that modulates the expression of many different genes.15,19,48–50 Nrf2 is a member of the cap ‘n’ collar (CNC) subfamily of basic region leucine zipper transcription factors and it mediates the induction of metabolizing and antioxidant enzymes by binding to antioxidant response elements (ARE). 51 In this study, we demonstrated that Nrf2 is significantly diminished in db/db mouse vessels and in HCSMCs cultured in HG. We found that the level of Nrf2 expression is important in regulating its downstream targets including NQO1 and BK-β1. The effect of Nrf2 in the regulation of BK-β1 protein expression appears to be mediated through its effects on MuRF1, which is repressed by Nrf2. We have recently reported that MuRF1 directly regulates BK-β1 expression and is responsible for the BK-β1 downregulation in diabetes and for BK channel dysfunction in type 1 diabetic mice. 13 This study is an extension of our previous findings, identifying Nrf2as the upstream regulator of MuRF1 expression in type 2 diabetic vessels.
Nrf2 is known to be neuroprotective 52 and cardioprotective 53 and activators of Nrf2 have been shown to be efficacious in the treatment of pathological conditions through its antioxidant and anti-inflammatory effects.21,54 Recently, DMF has been successfully employed in the treatment of human relapsing multiple sclerosis with favourable outcome and adverse effects profile.27,28 With treatment of diabetic mouse coronary arteries and isolated coronary arterial SMCs with this FDA-approved drug, we found that activation of Nrf2 preserved BK-β1 expression, restored vascular BK channel function and improved vascular SS-mediated vasodilation. These findings suggest that Nrf2 activation is effective in preventing diabetic-induced BK channelopathy and is a potential therapeutic target for the treatment of cardiovascular diseases related to diabetes.
In summary, this study confirms that type 2 diabetes is associated with vascular BK channelopathy due to reduced BK-β1 levels and loss of channel function from BK-β1 modulation. We found that the BK-β1 expression in diabetic vessels and cultured coronary SMCs is under the tight control of Nrf2 which is known to have antioxidant and anti-inflammatory function. We have also demonstrated that Nrf2 is deficient in diabetic vessels and in coronary SMCs exposed to HG. This Nrf2 deficiency leads to upregulation of MuRF1 and the augmented MuRF1 in turn targets BK-β1 for degradation, leading to a diminished level of expression with loss of BK-β1 modulation of BK channel function. An important finding of this study is that activation of Nrf2 results in restoration of BK-β1 expression, preservation of BK channel function and improved SS-mediated vasodilation in type 2 diabetes. Whether Nrf2 activation would be beneficial for the treatment of human diabetic vasculopathy will await the results of clinical trials.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This study was supported by grants from the National Institute of Health, National Institute of Heart Lung and Blood Institute (HL080118 and HL74180), the American Diabetes Association (ADA-JFA-07-39, ADA 1-12-BS-119 and ADA 1-16-IBS-195), the Natural Science Foundation of Jiangsu Province, China (BK20140249) and the National Natural Science Foundation of China (#81470489).