
Abstract
Aim:
We hypothesized that dipeptidyl peptidase (DPP)-4 inhibitor (vildagliptin) reduces fatal arrhythmias, cardiac dysfunction and infarct size caused by ischaemia–reperfusion (I/R) injury via its attenuation of cardiac mitochondrial dysfunction.
Methods:
In total, 26 rats were randomized to receive either 1 mL normal saline solution or 2.0 mg/kg vildagliptin intravenously (n = 13/group) 30 min prior to a 30-min left anterior descending coronary artery occlusion, followed by a 120-min reperfusion. Arrhythmia scores, cardiac functions, infarct size and mitochondrial function were evaluated.
Results:
Vildagliptin reduced the infarct size by 44% and mitigated cardiac dysfunction by preserving cardiac function without altering the incidence of cardiac arrhythmias. Vildagliptin increased expression of Bcl-2 and pro-caspase3 in the ischaemic area, whereas Bax and phosphorylated-connexin43/total-connexin43 were not altered. Vildagliptin attenuated cardiac mitochondrial dysfunction by reducing the reactive oxygen species level and mitochondrial swelling.
Conclusions:
DPP-4 inhibitor provides cardioprotection by reducing the infarct size and ameliorating cardiac dysfunction in I/R hearts by attenuating cardiac mitochondrial dysfunction and cardiomyocyte apoptosis.
Keywords
Introduction
Acute myocardial infarction is a major cause of morbidity and mortality in most nations. 1 Myocardial ischaemia is characterized by reduced blood flow to the heart muscle, leading to myocardial cell death. Interventions including percutaneous coronary intervention and thrombolysis have been used as reperfusion therapy. Although reperfusion is the definite myocardial salvage after ischaemia, reperfusion itself is also known to cause extended myocardial damage, that is, reperfusion injury. 2 It has been shown that ischaemia–reperfusion (I/R) injury contributes to adverse cardiovascular outcomes after myocardial ischaemia including infarct death and cardiac dysfunction. 2 Since type 2 diabetes mellitus (T2DM) patients have a higher risk for coronary heart disease 3 and have a worse prognosis after cardiovascular events, 4 and that several anti-diabetic drugs have been shown to cause undesired cardiac events, 5 currently used anti-diabetic drugs should address not only glycaemic control but also reduce the risk of cardiovascular events.
Dipeptidyl peptidase (DPP)-4 inhibitor, a novel anti-diabetic drug, has been used to treat T2DM in the past decade.6,7 DPP-4 inhibitor increased plasma glucagon-like peptide (GLP)-1 level by inhibiting the proteolytic enzyme DDP-4. 8 Besides improving glycaemic control, GLP-1 analogue and DPP-4 inhibitor have been shown to have cardioprotective effect in the ischaemic heart model in both preclinical and clinical settings.7,9–17 A recent study from our group demonstrated that treatment with vildagliptin 30 min prior to myocardial ischaemia insult could protect the heart by reducing infarct size and stabilizing cardiac electrophysiology during I/R injury. 9 However, the beneficial effects and the underlying protective mechanisms of vildagliptin during cardiac I/R injury are still unclear.
The purpose of this study was to investigate the cardioprotective effect of DPP-4 inhibitor, vildagliptin, on cardiac arrhythmia, cardiac function and infarct size and its underlying mechanisms in I/R rat hearts. We tested the hypothesis that vildagliptin reduces fatal arrhythmias, cardiac dysfunction and infarct size during I/R. To test the cardioprotective mechanisms, we determined the effects of vildagliptin on cardiac mitochondria and molecular aspects. We hypothesized that the cardioprotective mechanism of vildagliptin is via attenuating cardiac mitochondrial dysfunction and cardiomyocyte apoptosis.
Methods
Ethical approval
All experiments were approved by the Institutional Animal Care and Committee of the Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand. Adult male Wistar rats weighing 300–400 g were obtained from the National Animal Center, Salaya Campus, Mahidol University, Bangkok, Thailand. All the animals were housed in controlled temperature and humidity with 12-h light–dark cycle and fed with normal chow and water ad libitum before the study.
Surgical procedure of myocardial I/R
Rats were anesthetized by intramuscular injection of the combination of Zoletil® (50 mg/kg) and xylazine (3 mg/kg). 18 The animals were ventilated via tracheotomy with room air from the small animal ventilator (SAR-830 Series; CWE Inc., Ardmore, PA, USA) with a tidal volume of 300 cm3/min and at the rate of 53 breath/min. Lead II electrocardiogram (ECG) was monitored throughout the study. The left femoral vein was cannulated and used as a route for vehicle (saline) or drug administration. The right carotid artery was cannulated to measure left ventricular (LV) pressure and volume using a pressure–volume (P-V) conductance catheter (1.9F; Scisense Instrument, Ontario, Canada). The heart was exposed through a left thoracotomy at the fourth intercostal space. The left anterior descending coronary artery (LAD) was ligated by 5-0 silk suture at approximately 2 mm from its origin and threaded through a plastic snare to permit reversible occlusion of the coronary artery. 18 Myocardial ischaemia was confirmed by regional ischaemic pallor, ST elevation on the ECG and hypokinesia. Rats were subjected to 30 min of ischaemia, followed by 120 min of reperfusion.
Experimental protocol
In total, 26 male Wistar rats were randomly divided into two groups (n = 13 per group). The first group was assigned to receive 1.0 mL of normal saline solution and the second group to receive vildagliptin (2.0 mg/kg). Both normal saline solution and vildagliptin were administered intravenously at a rate of 0.33 mL/min 30 min prior to 30-min LAD occlusion, followed by 120-min reperfusion. ECG and cardiac function were recorded throughout the experiment. At the end of the protocol, the heart was re-occluded and excised for infarct size determination (n = 8/group), western blot analysis and cardiac mitochondrial function study (n = 5/group). Dosage of vildagliptin in this study, 2 mg/kg, has been shown to provide cardioprotection in our previous swine study. 9 According to He et al.’s study, 19 the plasma concentration of vildagliptin increased to a peak at 30 min after intravenous administration. The half-life of vildagliptin was 1.67 h after the peak, and the plasma level of vildagliptin can last for 10 h after administration. Therefore, vildagliptin drug level would be sufficiently effective throughout the entire experiment.
Cardiac function determination
Cardiac function was evaluated using a P-V catheter. The catheter was inserted into the right carotid artery and advanced into the LV chamber to record LV pressure and volume relationship.5,18 Heart rate (HR), left ventricular end-systolic pressure (LVESP), left ventricular end-diastolic pressure (LVEDP), maximal slope of the systolic pressure increment (+dP/dt), maximal slope of the diastolic pressure decrement (−dP/dt) and stroke volume (SV) were assessed at 15 min before the LAD occlusion, at the end of the ischaemic period and at the end of reperfusion using LabScribe2 program (iWorx Systems Inc., Dover, NH, USA).5,18
Arrhythmia determination
Lead II ECG was monitored throughout the experiment to record arrhythmias using PowerLab 4/25 (AD Instruments, Inc., Colorado Springs, CO, USA). Arrhythmias were characterized based on Lambeth Conventions 20 and scored as previously described by Curtis and Walker. 21 Arrhythmia score was classified into five levels as follows: 0 represented the number of premature ventricular contractions (PVCs) < 50 beats; 1 represented the number of PVC between 50 and 499 beats; 2 represented PVC > 500 beats and/or one episode of spontaneously reverting ventricular tachycardia or ventricular fibrillation (VT/VF); 3 represented more than one episode of spontaneously reverting VT/VF (<1 min of total combined duration); 4 represented 1–2 min of total combined VT/VF and 5 represented >2 min of VT/VF. The occurrence of VT/VF, time to first VT/VF and VT/VF duration were also determined.
Infarct size determination
At the end of each experiment, the heart was removed and mounted on the modified Langendorff apparatus via aorta. 18 The infarct size was defined by 0.5% Evans blue and 1.0% triphenyltetrazolium chloride (TTC) staining technique. 9 Evans blue was infused after LAD re-occlusion to evaluate the area at risk (AAR). After frozen overnight, the heart was cut perpendicularly from apex to the occlusion site into seven to eight slices. 18 Each slice was incubated in TTC for 12 min to discriminate the infarct tissue (white area) from the viable myocardium (red area). After fixation overnight with 4% paraformaldehyde, heart slices were photographed. Each area was measured using ImageTool software version 3.0.5,9,18
Western blot analysis
Heart tissues were collected from ischaemic and non-ischaemic (remote) areas, quickly frozen in liquid nitrogen and stored at −80°C until used as described previously. 22 A 100 mg of heart tissue was homogenized in 1 mL of extraction buffer (20 mmol/L Tris-HCl, 1 mmol/L Na3VO4, 5 mmol/L NaF, and cocktail protease and phosphatase inhibitor). The supernatant of tissue homogenate was collected after centrifugation (13,000 r/min, 10 min at 4°C) and defined as total protein. The total protein was mixed with loading buffer [10% mercaptoethanol, 0.05% bromophenol blue, 75 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulphate (SDS) and 10% glycerol] and heated at 95°C for 10 min. Then, the protein samples were subjected to 10% or 15% SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes using a semi-dry transfer system (Trans-Blot SD; Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% non-fat skimmed milk in TBST (20 mM Tris-HCl, pH 7.6, 137 mM NaCl and 0.1% Tween-20) for 1 h and incubated with specific antibodies against Bax, Bcl-2, pro-caspase3, phosphorylated connexin43 (P-Cx43) (S368), total connexin43 (T-Cx43) and β-actin (Cell Signaling Technology) overnight at 4°C. On the next day, the membranes were washed and incubated with horseradish peroxidase–conjugated secondary antibody (Santa Cruz Biotechnology, Inc., CA, USA) for 1 h at room temperature. The signals were developed by incubating with enhanced chemiluminescense reagent and subjected to autoradiography. The immunoblot films were scanned and the band density was analysed using ImageJ analysis (NIH image).
Cardiac mitochondrial function study
At the end of each experiment, cardiac mitochondria were isolated from both ischaemic and remote areas. 5 Cardiac mitochondrial function including cardiac mitochondrial reactive oxygen species (ROS) production, cardiac mitochondrial membrane potential change (ΔΨM) and cardiac mitochondrial swelling were determined as previously described.5,9,23 In brief, dichlorohydrofluorescein diacetate dye was used to determine the ROS production in cardiac mitochondria. The fluorescence intensity was determined at an excitation wavelength of 485 nm and emission wavelength of 530 nm. The dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) was used to determine the change in the mitochondrial membrane potential. JC-1 monomer (green fluorescence) was excited at 485 nm and the emission detected at 530 nm. JC-1 aggregate form (red fluorescence) was excited at 485 nm and the emission detected at 590 nm. Cardiac mitochondrial depolarization was indicated by a decrease in the red/green fluorescence intensity ratio. Cardiac mitochondrial swelling was indicated by the decrease in absorbance of mitochondrial suspension at 540 nm wavelength.
Cardiac mitochondria morphology
Electron microscopy was used to determine the morphology of cardiac mitochondria. Isolated mitochondria from both ischaemic and remote areas were fixed overnight by mixing 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, at 4°C. Then, the pellet was postfixed in 1% cacodylate-buffered osmium tetroxide for 2 h at room temperature. The pellets were dehydrated in a graded series of ethanol and embedded in Epon–Araldite and were cut by a diamond knife into ultra-thin sections (60–80 nm thick), placed on copper grids and stained with the combination of uranyl acetate and lead citrate. Mitochondrial morphology was observed with a transmission electron microscope. 24
Statistical analysis
Data were presented as mean ± standard error (SE). Statistical comparison of variables at baseline, at the end of ischaemia and at the end of reperfusion (within group) was analysed with Wilcoxon signed-rank test. Comparison of infarct size, arrhythmia parameters and protein expression (among groups) was performed using Mann–Whitney U test. Chi-square test was performed to compare VT/VF incidence. All statistical analyses were performed with SPSS version 10.0; p < 0.05 was considered as statistically significant difference.
Results
Vildagliptin reduced infarct size in I/R hearts
In this study, we found that the AAR was not significantly different between the saline- and vildagliptin-treated groups (39.0% ± 5.8% vs 38.7% ± 3.5%). However, the infarct size in the vildagliptin-treated group was markedly smaller than the saline-treated group, accounting for an approximately 44% reduction (Figure 1(a)). The Bcl-2, an anti-apoptotic marker, was significantly increased in the vildagliptin-treated group in ventricular tissues in both remote and ischaemic areas (Figure 1(b)). However, Bax expression level was not different between groups (Figure 1(c)). The pro-caspase3 level in the ischaemic area was also increased in vildagliptin-treated group (Figure 1(d)).
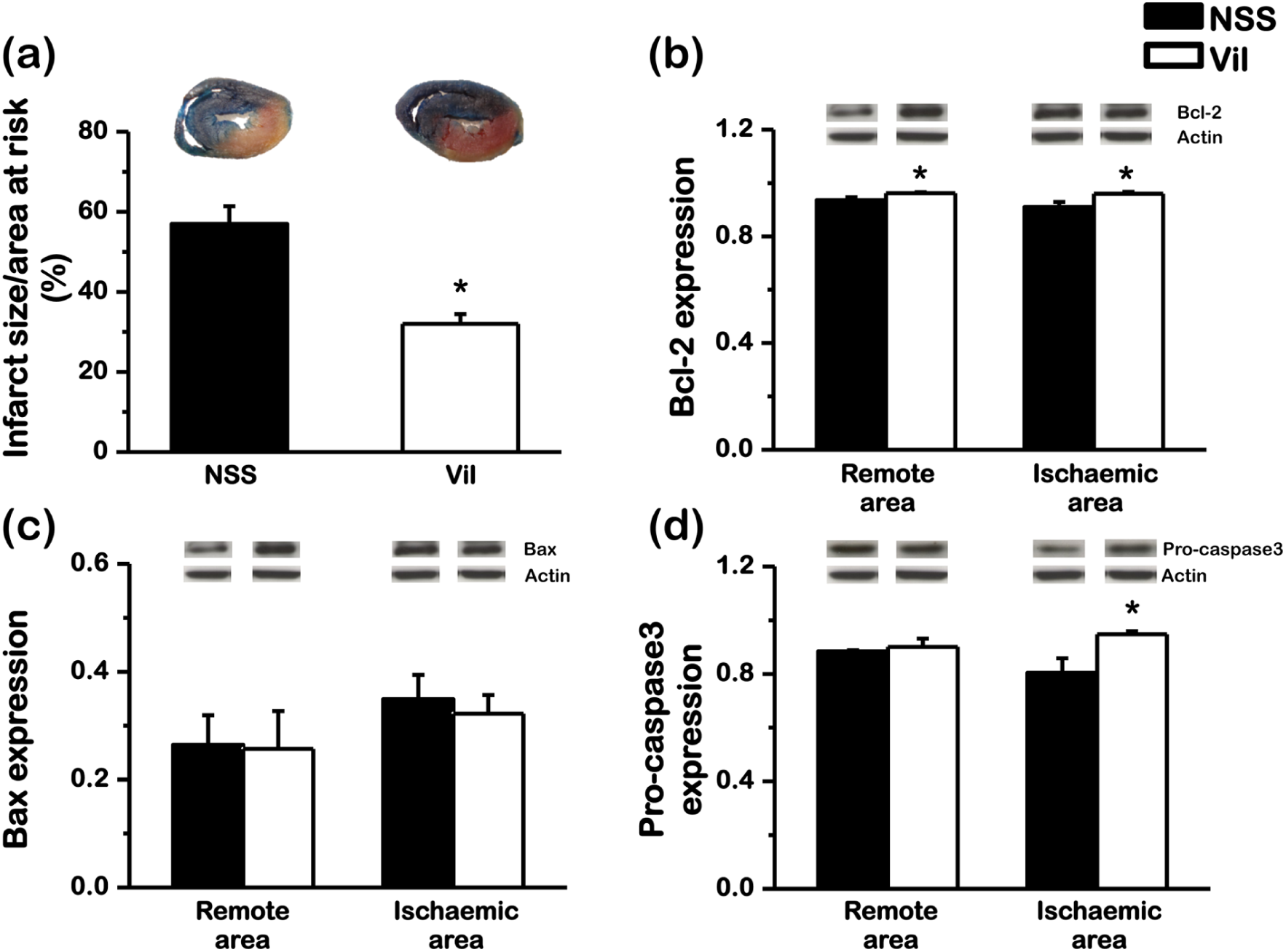
Effect of vildagliptin on infarct size, anti-apoptotic and pro-apoptotic cascades. The area at risk was not different between the vildagliptin- and normal saline-treated groups. (a) Vildagliptin markedly reduced the infarct size by 44%, compared with the normal saline-treated group. Vildagliptin significantly increased (b) Bcl-2 and (d) pro-caspase3 expression in ischaemic area, but had no effect on (c) Bax expression (*p < 0.05 versus NSS).
Occurrence of arrhythmias
The occurrence of VT/VF during both the ischaemic and reperfusion periods was not different between vildagliptin-treated group and saline-treated group (Figure 2(a)). Furthermore, vildagliptin neither prolonged the time interval from ischaemia insult to the fatal arrhythmia (Figure 2(b)) nor decreased arrhythmia scores (Figure 3(a)), compared to the saline-treated group. The corrected QT interval was not different between normal saline- and vildagliptin-treated groups (176 ± 10 vs 185 ± 8 ms, p = 0.565). Western blot analysis of Cx43 demonstrated that the level of phosphorylated Cx43 per total Cx43 (P-Cx43/T-Cx43) was not different between vildagliptin-treated group and saline-treated group (Figure 3(b)).
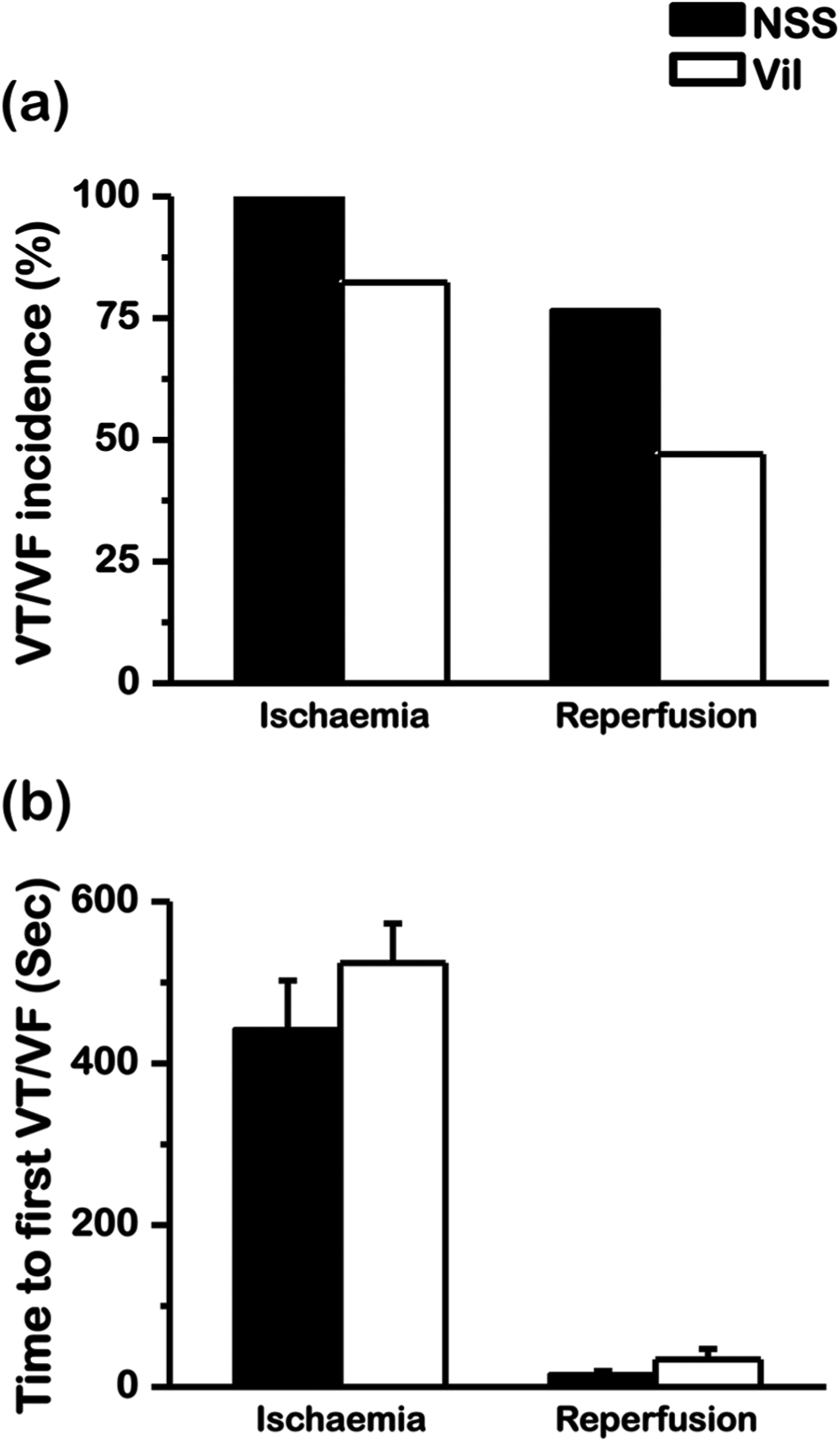
Vildagliptin and the occurrence of arrhythmia. Vildagliptin did not reduce (a) VT/VF incidence or (b) time to first VT/VF onset.
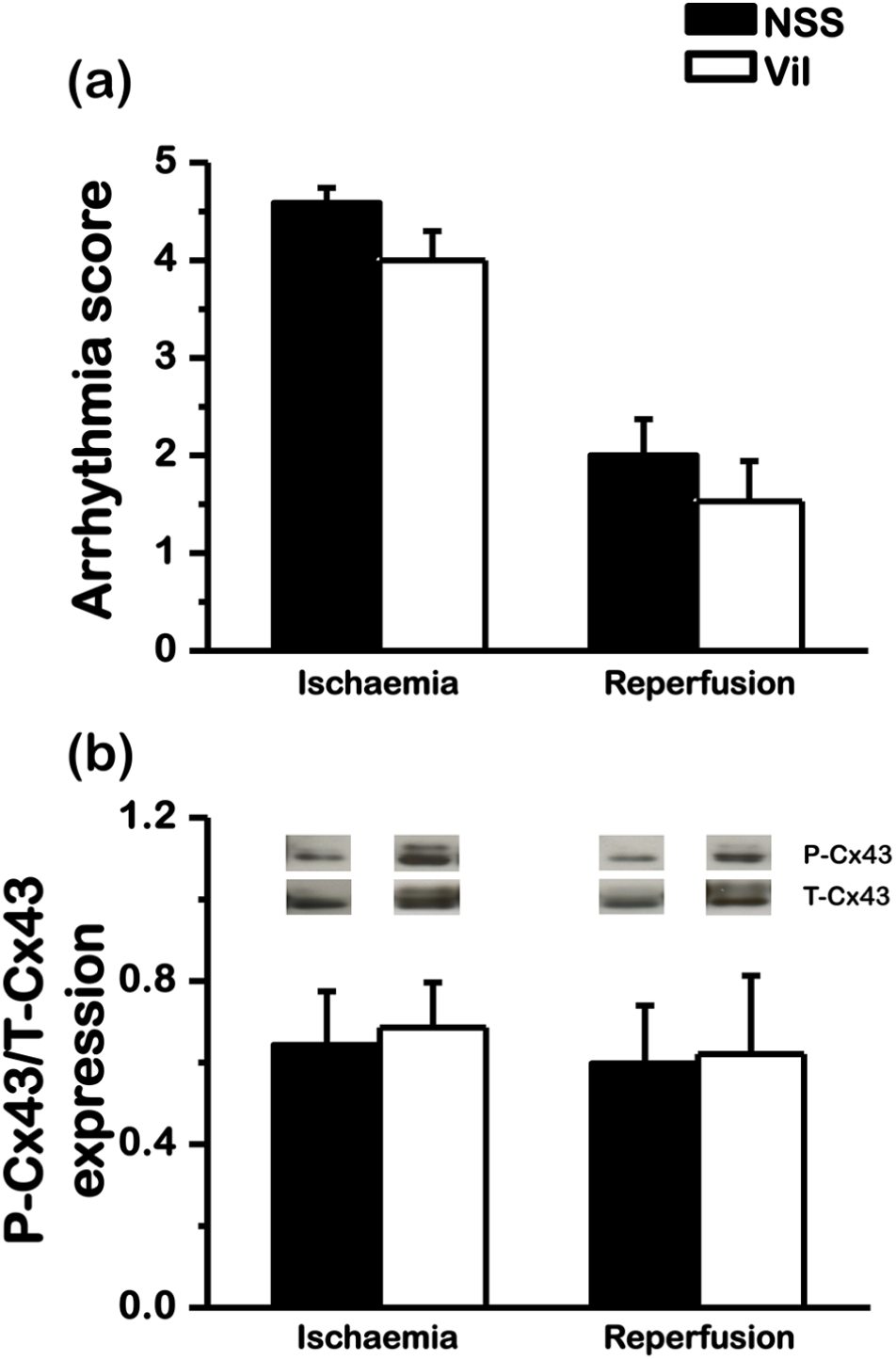
Effect of vildagliptin on arrhythmogenesis. (a) Arrhythmia score was not different between vildagliptin-treated group and saline-treated group, which is consistent with the (b) unaltered P-Cx43/T-Cx43 expression in vildagliptin-treated group, compared to saline-treated group.
Vildagliptin preserved systolic function and SV
The changes in LV function parameters at different time points are shown in Table 1. During the ischaemic period, saline-treated rats demonstrated both systolic and diastolic dysfunction as indicated by significantly decreased LVESP, +dP/dt and SV and significantly increased LVEDP and −dP/dt, compared to the baseline. On the other hand, vildagliptin-treated rats had better cardiac performance during the ischaemic period in which LVESP, +dP/dt and SV during ischaemia were improved and were not different from those at the baseline. However, treatment with vildagliptin could not alleviate diastolic dysfunction during the ischaemic period. During the reperfusion, cardiac function was restored back to the baseline in vildagliptin-treated group.
Effects of vildagliptin on cardiac functions.
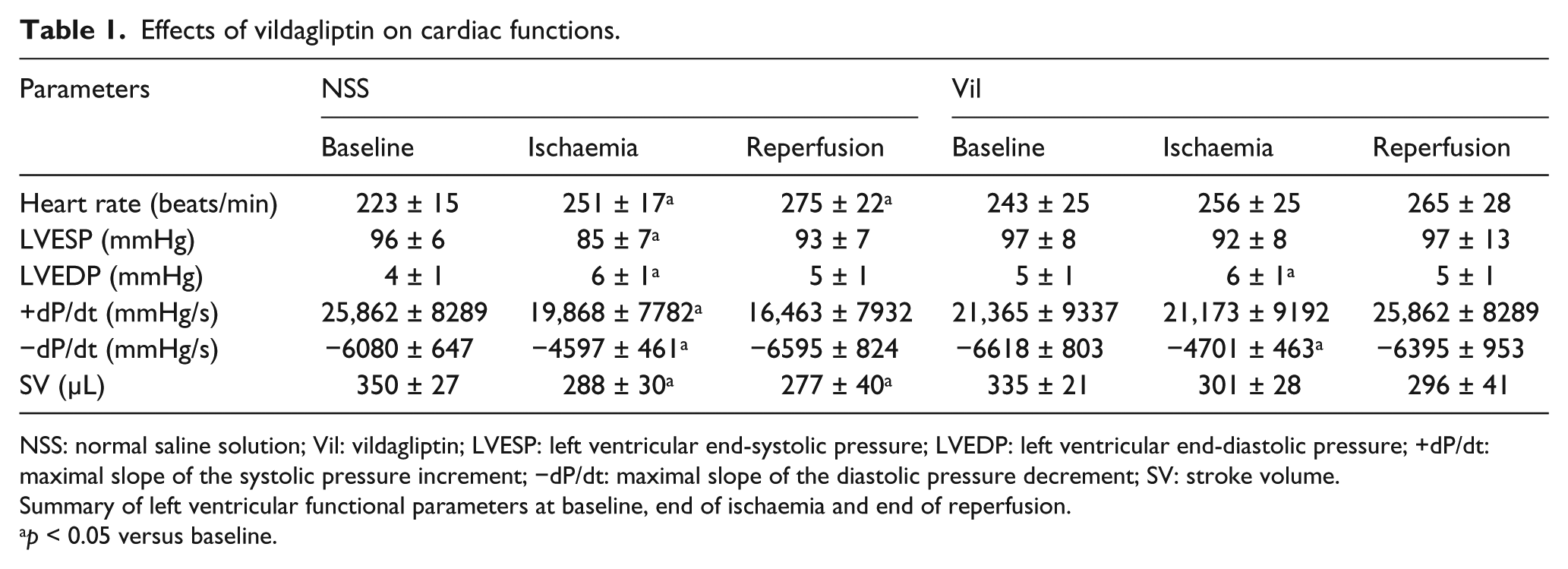
NSS: normal saline solution; Vil: vildagliptin; LVESP: left ventricular end-systolic pressure; LVEDP: left ventricular end-diastolic pressure; +dP/dt: maximal slope of the systolic pressure increment; −dP/dt: maximal slope of the diastolic pressure decrement; SV: stroke volume.
Summary of left ventricular functional parameters at baseline, end of ischaemia and end of reperfusion.
p < 0.05 versus baseline.
Vildagliptin mitigated cardiac mitochondrial dysfunction
Cardiac mitochondria taken from the ischaemic myocardium were characterized by increased mitochondrial ROS production (Figure 4(a)), decreased absorbance which indicated mitochondrial swelling (Figure 4(b)) and decreased the red/green fluorescent intensity ratio which indicated mitochondrial depolarization (Figure 4(c)), compared to those from the non-ischaemic area. Treatment with vildagliptin improved cardiac mitochondrial function in the ischaemic myocardium, as indicated by significantly decreased ROS level (Figure 4(a)) and mitochondrial swelling (Figure 4(b)). The total ROS level was also significantly higher in the control group than in the vildagliptin-treated group. However, vildagliptin did not attenuate mitochondrial depolarization (Figure 4(c)). Representative electron microscope pictures of cardiac mitochondria are shown in Figure 4(d). In the saline group, abnormal morphology of cardiac mitochondrion in the ischaemic area can be seen together with the loss of cristae, indicating mitochondrial swelling. Vildagliptin treatment markedly reduced mitochondrial swelling in the ischaemic area caused by I/R injury.
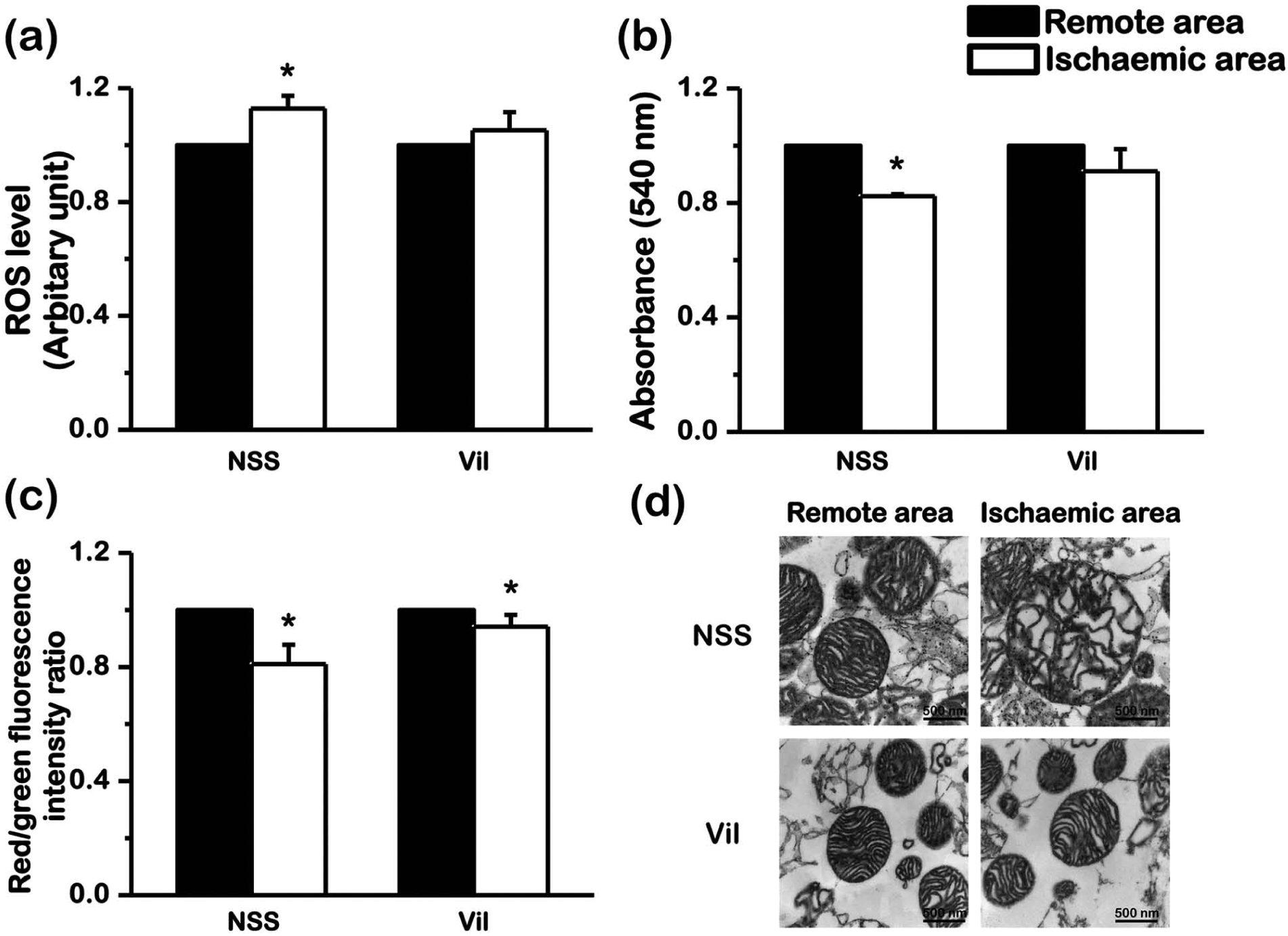
Effect of vildagliptin on cardiac mitochondria after ischaemia–reperfusion period. Vildagliptin improved cardiac mitochondrial function by (a) reducing ROS production and (b) attenuating mitochondrial swelling in ischaemic area. However, (c) vildagliptin did not attenuate mitochondrial membrane depolarization. (d) Vildagliptin also attenuated the deterioration of cardiac mitochondrial morphology (*p < 0.05 versus remote area).
Discussion
In this study, we determined the cardioprotective effects of vildagliptin and its underlying mechanism in I/R rat hearts. The major findings of this study are as follows. Acute treatment with vildagliptin provided cardioprotective effects during I/R insult by (1) markedly decreasing the infarct size; (2) improving cardiac function by preserved systolic function and SV; (3) significantly increasing expression of Bcl-2 and pro-caspase3 in ischaemic myocardium, without altering the Bax expression and (4) preventing cardiac mitochondrial dysfunction caused by I/R injury by reducing ROS production and attenuating mitochondrial swelling, without altering mitochondrial membrane depolarization. However, vildagliptin neither attenuated cardiac arrhythmias occurring during I/R nor altered the P-Cx43/T-Cx43 ratio.
The size of infarct myocardium caused by myocardial ischaemia has been shown to be associated with the severity of LV dysfunction as well as mortality.25,26 Therefore, interventions to reduce the infarct size have been extensively investigated for decades.2,9,13,25,27,28 Previous studies demonstrated that decreased mortality and improved LV function were associated with a smaller infarct size.25,26,29 In this study, we demonstrated that acute administration of vildagliptin markedly reduced the infarct size caused by I/R injury by 44%. This infarct limiting effect has been observed in previous in vivo studies in genetic deletion or pharmacological inhibition of DDP-4 activity.9,10,14,15 Since myocardial infarction plays an essential role in cardiac dysfunction, reduction in infarct size would provide a benefit on contractility. We found that vildagliptin improved LV systolic function and SV in hearts subjected to I/R, while it failed to improve diastolic function. These findings were consistent with a previous study using DPP-4 deleted rat subjected to 45-min occlusion followed by 120-min reperfusion. 15 The mechanism by which the drug failed to improve diastolic function is still not fully understood. Interestingly, the infarct size was dramatically reduced ranging from 38% to 64% in rats and 17% in pigs.9,10,14,15 Although acute administration of DPP-4 inhibitor has been shown to reduce the infarct size,9,10,14,15 several studies reported that prolonged treatment with DPP-4 inhibitors failed to attenuate infarct size, but improved survival rate.27,30,31 These discrepancies could be due to the differences in time and duration of drug administration as well as different species and ischaemia models used in those studies.
Although reperfusion is the means to relieve myocardial damage from necrosis and limit infarct size from myocardial ischaemia, it paradoxically promoted extended myocardial injury due to myocardial apoptosis.2,32 Bax and Bcl-2 have been shown to regulate outer mitochondrial membrane pore formation, which involves in apoptotic process. 33 Once cytochrome C is released from the pores, caspase cascade proteins such as caspase9 and caspase3 are activated and set apoptosis in motion.33,34 Bax is a pro-apoptotic protein-promoting apoptosis, whereas Bcl-2 is an anti-apoptotic protein-preventing apoptosis. Our study demonstrated that vildagliptin alleviated myocardial apoptosis by increasing anti-apoptotic protein Bcl-2 causing the less activated caspase3, thus resulting in higher level of pro-caspase3.
Mitochondria also play pivotal roles in myocardial survival or death following ischaemia and reperfusion.35,36 The burst of ROS level during I/R 37 period caused mitochondrial permeability transition pore (mPTP) opening, 36 resulting in dissipating of adenosine triphosphate (ATP) and mitochondrial swelling, 38 which subsequently lead to an activation of the apoptotic pathway and eventually cardiac cell death.35,36,39 Moreover, the increased ROS level also causes mitochondrial membrane depolarization which has been shown to be responsible for cardiac arrhythmia. 40 In this study, we demonstrated the first time protective effect of vildagliptin on cardiac mitochondria during I/R. We found that vildagliptin reduced ROS level and mitochondrial swelling and could well preserve cardiac mitochondrial morphology. This could also play an important role in infarct size reduction.
Since fatal arrhythmias are responsible for the major cause of death in acute myocardial infarction patients, pharmacological intervention to reduce these arrhythmias would be a therapeutic target to reduce mortality. In this study, we demonstrated the first time effects of DPP-4 inhibitor on cardiac arrhythmia in rats. Vildagliptin failed to reduce fatal cardiac arrhythmia during I/R. Treatment with vildagliptin could not reduce VT/VF incidence, arrhythmia score and time interval to the onset of fatal arrhythmia, which is consistent with our previous study in a clinically relevant pig model. 9 Previous studies demonstrated that Cx43, a major cardiac gap junction in the ventricles, plays a crucial role in facilitating the propagation of electrical impulse from cell to cell, allowing the heart to contract synchronously.41,42 The reduction of Cx43 expression as well as the alteration of phosphorylation and dephosphorylation at different serine residual of Cx43 has been shown to impair the propagation of electrical impulses and promote re-entrant arrhythmia.43–45 Moreover, increased phosphorylation of Cx43 at serine 368 has been shown to protect arrhythmias from the ischaemic injury. 46 Our findings that the level of P-Cx43/T-Cx43 in vildagliptin-treated myocardium was not different from that in the saline-treated group could explain the results that vildagliptin did not attenuate arrhythmias induced by I/R in this study.
Besides Cx43, cardiac mitochondrial membrane depolarization has been shown to associate with cardiac arrhythmias. I/R event has been shown to produce high oxidative stress, for example, ROS. When the burst of ROS reaches threshold, it can induce ROS release in neighbouring mitochondria through the mitochondrial network, thus affecting the whole cell.37,47 Oxidative stress is a major pathophysiological route to cause a collapse of mitochondrial membrane potential, resulting in the impairment of mitochondrial function, including ATP production. 48 The collapse of mitochondrial membrane potential (i.e. mitochondrial depolarization) leads to an activation of sarcolemmal KATP channels, resulting in spatiotemporal heterogeneity of action potentials, leading to a conduction block, inducing re-entry and subsequently initiating VT or VF. 40 In this study, vildagliptin did not improve cardiac mitochondrial membrane depolarization caused by I/R injury. This could be another mechanistic insight regarding the findings that vildagliptin could not attenuate arrhythmia in this study.
Our study limitation is that although we demonstrated that vildagliptin exerted infarct limiting effect and improved cardiac performance during I/R, the histopathology as well as GLP-1 level and other inflammatory cytokines which could play a role for cardioprotection of vildagliptin treatment were not determined.
Conclusion
DPP-4 inhibitor provides cardioprotection by reducing the infarct size and ameliorating LV dysfunction in I/R rat hearts by attenuating cardiac mitochondrial dysfunction and cardiomyocyte apoptosis. These findings provide important information regarding cardiac benefits of this DPP-4 inhibitor in addition to its glycaemic control. Future clinical studies are needed to determine its clinical usefulness in patients with acute myocardial infarction.
Footnotes
Declaration of conflicting interests
None.
Funding
This study was supported by grants from the Thailand Research Fund (RTA5580006 to Nipon Chattipakorn; BRG5480003 to Siriporn Chattipakorn), the Thailand Research Fund’s Royal Golden Jubilee PhD program (Nipon Chattipakorn and Kroekkiat Chinda) and a Chiang Mai University Excellent Center Award (Nipon Chattipakorn).