
Abstract
Background
Diabetic retinopathy (DR), a leading cause of vision loss, arises from chronic inflammation, vascular injury, and immune dysregulation. Aging-related mechanisms, including inflammaging, further exacerbate immune activation. The renin–angiotensin-aldosterone system (RAS) has been implicated in vascular dysfunction and chronic inflammation in DR. Paxillin (PXN), a cytoskeletal adaptor regulating immune cell migration, has not been fully explored in the context of DR and aging-related immune responses.
Objective
To determine how PXN+ monocytes contribute to DR progression, with a focus on SELPLG-SELL-mediated chemotaxis, PXN-RAS interactions, and age-related inflammatory remodeling.
Methods
Mendelian randomization (MR) and single-cell transcriptomics identified PXN as a putative driver of DR. Functional assays were performed in RAW264.7 macrophages with PXN overexpression or knockdown under high-glucose conditions, with or without Losartan treatment. qPCR, CCK-8, ELISA, flow cytometry, and immunoblotting were used to assess inflammatory and oxidative pathways.
Results
MR revealed a causal association between PXN and DR risk, highlighting the SELPLG-SELL axis as a major chemotaxis pathway. Single-cell RNA-seq showed PXN+ monocytes amplify T/NK cell crosstalk and immune infiltration. PXN overexpression promoted M1 polarization, upregulated SELL/SELPLG, increased IL-6, IL-17, and IL-23 secretion, and enhanced ACE-driven oxidative stress, attenuating Losartan's anti-inflammatory effects. These findings link PXN+ monocytes to inflammaging-like immune remodeling in DR.
Conclusions
PXN amplifies pro-inflammatory chemotaxis and immune aging in DR, undermining RAS inhibition. Targeting PXN may mitigate inflammaging-driven retinal inflammation and improve therapeutic responses in diabetic retinopathy.
This study identifies PXN+ monocytes as pivotal drivers of immune dysregulation and therapeutic resistance in diabetic retinopathy (DR), a microvascular complication closely linked to renin–angiotensin system (RAS) activity. PXN promotes Th1 polarization and recruits MKI67+ T/NK cells via the SELPLG–SELL axis, amplifying retinal inflammation through TNF-α, IFN-γ, and IL-2. It also induces M1 macrophage polarization, upregulates ACE, and increases oxidative stress. Notably, PXN overexpression abrogates the anti-inflammatory effects of the RAS inhibitor Losartan, revealing a novel mechanism of RAS blockade resistance. By integrating Mendelian randomization, single-cell RNA sequencing, and in vitro experiments, we establish PXN as a central immunomodulator, highlighting its therapeutic potential in DR and other RAS-driven inflammatory diseases.
Keywords
Background
Diabetic retinopathy (DR) is a leading microvascular complication of diabetes mellitus (DM) and one of the primary causes of vision loss in working-age adults globally.1,2 In China, the prevalence of diabetes ranges from 24.7% to 27.3%, with a concomitant rise in the incidence of DR.3,4 The risk of DR increases with the duration of DM, underscoring the urgent need for effective prevention and management strategies.5–8 DR pathogenesis involves multiple processes, including hypoxic-ischemic injury, oxidative stress, inflammation, immune activation, and vascular dysfunction.9–12 A growing body of evidence highlights immune-mediated mechanisms as central to the disease's progression, especially in the context of aging.13,14
As individuals age, the efficiency of immune responses declines, and this immunosenescence contributes to an enhanced inflammatory state that exacerbates DR.13,14 In the retina, monocyte, macrophage, and T cell recruitment and activation contribute to vascular injury through the secretion of pro-inflammatory cytokines such as TNF-α, IL-6, and MCP-1.15–17 These cytokines amplify local inflammation and create a self-perpetuating loop of immune cell recruitment, promoting further retinal damage and aggravating disease severity. Macrophage infiltration has been closely correlated with DR progression, with pro-inflammatory cytokines such as TNF-α and IL-1β increasing vascular permeability and driving neovascularization.18–21 T cell infiltration, particularly in the aging retina, also exacerbates microvascular damage, leading to vision impairment in both type 1 and type 2 diabetes.17,22
Oxidative stress, a hallmark of aging, further intensifies immune cell chemotaxis in the diabetic retina.23,24 Hyperglycemia-induced reactive oxygen species (ROS) activate signaling pathways that promote immune cell adhesion and migration, accelerating retinal inflammation and vascular damage. 25 The recruited immune cells interact with retinal endothelial cells and pericytes, disrupting the blood-retina barrier, contributing to macular edema, and driving neovascularization.26,27 These interactions are central to the vascular injury observed in DR and are further exacerbated by aging-related changes in immune cell function and tissue repair mechanisms.12,28
Recent studies have implicated Paxillin (PXN), a cytoskeletal adaptor protein, in regulating immune cell activation and vascular dysfunction in DR. PXN modulates monocyte/macrophage activation and T cell differentiation, contributing to the immune dysregulation and vascular damage seen in DR, especially in the context of aging.29,30 The molecular mechanisms of immune cell-mediated injury in DR are complex and involve multiple signaling networks, including the regulation of matrix metalloproteinases (MMPs) that degrade the extracellular matrix and compromise vascular integrity.31,32 These processes are further amplified by aging, which affects immune responses and exacerbates inflammation in the retina.
In light of the central role of immune activation and chemotaxis in DR pathogenesis, targeting these processes holds promise as a therapeutic strategy.17,33 Modulating immune cell recruitment and function could interrupt the inflammation-vascular injury cycle, thereby slowing disease progression and alleviating the impact of aging on retinal health. Advances in genomics and transcriptomics offer new insights into the molecular mediators of immune-driven DR, enabling the identification of key genes involved in retinal inflammation and vasculopathy.34–37 By integrating Mendelian randomization (MR), single-cell RNA sequencing, and transcriptomic analyses, this study aims to identify chemotaxis-related immune genes implicated in DR, providing a foundation for targeted immunomodulatory therapies to address this unmet clinical need in aging populations.38–41
Methods
Causal relationship between plasma proteins and diabetic retinopathy: MR analysis
We assessed the causal effects of plasma proteins on DR using Mendelian randomization (MR).42–46 Instrumental variables (IVs) were required to meet three assumptions: (i) association with the exposure of interest; (ii) independence from confounders; and (iii) influence on the outcome solely via the exposure. IVs were obtained from publicly available plasma protein genome-wide association studies (GWAS) based on genome-wide significance (P < 5 × 10−8). Independent single nucleotide polymorphisms (SNPs) were selected via linkage disequilibrium (LD) clumping using 1000 Genomes Project Phase 3 (EUR) data. Datasets were harmonized using the harmonise_data function in R, removing SNPs with intermediate allele frequencies; missing outcome SNPs were not replaced with proxies. Five MR approaches were applied: inverse variance weighting (IVW), MR-Egger, weighted median, simple mode, and weighted mode. IVW was the primary method due to its estimation efficiency, contingent on the absence of horizontal pleiotropy. MR-Egger, which relies on the Instrument Strength Independent of Direct Effect (InSIDE) assumption, was used to assess pleiotropy via the intercept term. MR-PRESSO was applied to detect and correct for pleiotropy, while Cochran's Q statistic evaluated heterogeneity. Sensitivity analyses were conducted using the leave-one-out method.
Identification and validation of key genes
Aging-related genes with a relevance score >5 were retrieved from the GeneCards database. GEO datasets GSE221521 and GSE234447 were obtained for transcriptomic analysis, with GSE221521 as the training cohort and GSE234447 as the validation cohort. Following the analytical approach described previously, differentially expressed genes (DEGs) were identified using the limma package, with particular attention to genes upregulated in DR.47–50 Overlapping DR-upregulated genes, aging-related genes, and MR-derived candidates yielded key genes, among which PXN was identified and validated in GSE234447.
Correlation analysis of key genes
In GSE221521, Pearson correlation analysis identified genes positively correlated with PXN. Protein–protein interaction (PPI) networks were constructed using STRING to map proteins associated with PXN, and a PXN-centered PPI network was generated to explore potential direct interactors.
Functional and immune infiltration analysis
GO and KEGG enrichment analyses were performed using the clusterProfiler package for plasma proteins associated with DR from MR analysis, as well as for PXN and its correlated genes. For immune infiltration analysis, GSE221521 samples were stratified into high- and low-PXN groups based on median expression. The CIBERSORT algorithm quantified the relative abundance of 22 immune cell types, and differences between groups were assessed. Spearman correlation tested associations between PXN expression and immune cell proportions. Immune pathway activity was evaluated using single-sample gene set enrichment analysis (ssGSEA), while immune, stromal, and ESTIMATE scores were calculated with the estimate package to compare tumor microenvironment characteristics between PXN expression groups.
Single-cell analysis
Single-cell RNA sequencing (scRNA-seq) data from GSE248284 were processed using the Seurat package, with tidyverse and ggplot2 for enhanced visualization.51–54 Standard preprocessing steps included normalization (NormalizeData), variable feature selection (FindVariableFeatures), and scaling (ScaleData). Batch effects between DR and non-diabetic retinopathy (NDR) samples were corrected using the Harmony package. T cells, NK cells, and mononuclear phagocytes (MPs) were re-clustered based on canonical biomarkers. Classical monocytes were further subdivided into PXN− and PXN+ subsets according to PXN expression. Intercellular communication networks were inferred using CellChat and visualized with the patchwork package.
Cell culture
RAW264.7 murine macrophages (ATCC, Rockville, MD, USA) were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco) containing 10% heat-inactivated fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin, at 37 °C in a humidified 5% CO₂ incubator. For pharmacological treatments, Losartan (≥99% purity, #61188) and lipopolysaccharide (LPS, E. coli O111:B4, #L2630) (Sigma-Aldrich, St Louis, MO, USA) were applied at the concentrations indicated in figure legends.
Lentiviral overexpression and knockdown of PXN
For gain- and loss-of-function studies, RAW264.7 cells were transduced with lentiviral vectors encoding full-length murine PXN (OE-PXN) or short hairpin RNAs targeting PXN (shPXN-1, shPXN-2) (Open Biosystems), in the presence of 8 μg/mL polybrene for 24 h. Non-targeting vectors served as negative controls (NC and sh-NC). After transduction, cells were selected with 2 μg/mL puromycin for 72 h to enrich for positively transduced populations.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen) and reverse-transcribed with a cDNA synthesis kit (Thermo Fisher Scientific). qRT-PCR was performed using SYBR Green Master Mix (Takara) on a LightCycler 480 system (Roche), with GAPDH as an internal control. Relative expression was calculated using the 2−ΔΔCt method. Primer specificity and efficiency were validated prior to use, with an annealing temperature of 60 °C.
Cell viability assay (CCK-8)
Cell viability was assessed using the Cell Counting Kit-8 (Dojindo). Briefly, 1 × 10⁴ cells/well were seeded in 96-well plates, treated for 24 h, followed by addition of 10 μL CCK-8 reagent. After 2 h incubation at 37 °C, absorbance at 450 nm was measured using a microplate reader (Bio-Rad). 55
Flow cytometry
For surface marker analysis, cells were stained with fluorophore-conjugated antibodies (BioLegend) for 30 min at 4 °C in the dark. Intracellular ROS levels were determined using DCFH-DA (10 μM; Beyotime) for 30 min at 37 °C. Samples were analyzed on a BD FACSCanto II flow cytometer, and data were processed with FlowJo v10. 56
ELISA
Cell culture supernatants collected after 24 h stimulation were analyzed for IL-6, IL-17, and IL-23 using ELISA kits (MultiSciences, Hangzhou, China) according to the manufacturer's instructions. Absorbance was read at 450 nm.
Western blotting
Cells were lysed in RIPA buffer with protease inhibitors, and protein concentrations determined by BCA assay (Beyotime). Equal protein amounts were separated via SDS-PAGE, transferred to PVDF membranes, and blocked in 5% non-fat milk. Membranes were incubated overnight at 4 °C with anti-ACE (Abcam) primary antibodies, followed by HRP-conjugated secondary antibodies. Signals were detected using an ECL kit (Thermo Scientific) and quantified with ImageJ.57,58
Immunofluorescence staining
RAW264.7 cells grown on coverslips were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 10% BSA. Primary antibodies against ACE, CD86, CD163, IL-1β, IL-6, or TNF-α (Abcam) were applied overnight at 4 °C, followed by Alexa Fluor-conjugated secondary antibodies. Nuclei were counterstained with DAPI, and images captured using an Olympus fluorescence microscope.
High glucose and losartan treatment
To mimic diabetic conditions, RAW264.7 cells were exposed to high glucose (25 mM D-glucose; Sigma-Aldrich) for 24 h. Where indicated, Losartan (10 μM; Sigma-Aldrich) was added 1 h prior to high glucose stimulation. OE-PXN transduction was performed 48 h before drug treatment.
Statistical analysis
All experiments were performed in triplicate. Data are presented as mean ± standard deviation. Statistical analyses were conducted using GraphPad Prism 8.0.1. For multiple groups, one-way ANOVA with Tukey's post hoc test was used; for two-group comparisons, an unpaired Student's t-test was applied. Data normality and variance homogeneity were verified prior to analysis. A p-value <0.05 was considered statistically significant.
Results
Causal associations between plasma proteins and diabetic retinopathy identified via Mendelian randomization
To evaluate the robustness of plasma protein–DR associations, a leave-one-out sensitivity analysis was performed using multiple SNPs as instrumental variables. In each panel of Supplementary Figure 1, red dots indicate recalculated MR estimates after sequential SNP removal, black dots represent individual SNP effect sizes, and horizontal bars denote 95% confidence intervals. MR estimates remained stable across all iterations (Figure 1A), indicating no single SNP disproportionately influenced the outcome and minimizing pleiotropic bias. Based on this validated framework, we conducted two-sample MR using the inverse-variance weighted (IVW) method, supported by MR-Egger and weighted median models. Twelve plasma proteins showed significant causal associations with DR (Figure 2), with consistent results across methods, suggesting that genetically predicted levels of these proteins may contribute directly to disease development. These findings provide a solid foundation for downstream transcriptomic and cellular investigations.

Functional characterization of dr-associated plasma proteins identified by mr. (A) MR tree plots illustrating SNP effects on protein levels (x-axis) and DR risk (y-axis), with significant SNPs marked in red. (B) Gene Ontology (GO) enrichment analysis showing overrepresented biological processes, molecular functions, and cellular components for DR-associated proteins, with significance indicated by adjusted p-values. (C) KEGG pathway enrichment analysis highlighting key pathways, ranked by significance. (D) Protein–protein interaction (PPI) network of DR-associated proteins, with highly connected clusters indicating potential mechanistic pathways.

Mendelian randomization analysis between plasma proteins and diabetic retinopathy. Scatterplots depict the causal associations between genetically predicted plasma protein levels (x-axis, SD units) and DR risk (y-axis, log odds) using the inverse-variance weighted (IVW) method, with MR-Egger and weighted median results shown for sensitivity analyses (blue: IVW, green: MR-Egger). Error bars represent 95% CIs, and heterogeneity was assessed via Cochran's Q test. Proteins with significant associations after multiple testing correction are highlighted, supporting potential causal roles in DR while accounting for pleiotropy (MR-Egger intercept).
Functional annotation and transcriptomic prioritization identify PXN as a central regulator in diabetic retinopathy
To define the biological relevance of MR-identified proteins, we performed functional enrichment analysis on 76 candidates. GO analysis indicated significant enrichment in processes related to programmed cell death, leukocyte activation, and immune responses (Figure 1B), while KEGG analysis highlighted TNF-α signaling and MMP activation pathways (Figure 1C), both implicated in retinal inflammation and vascular remodeling. PPI network construction identified TNF-α– and MMP-related molecules as key hubs (Figure 1D), underscoring inflammatory and matrix-degrading drivers of DR pathology. Integrating transcriptomic data from the GSE221521 dataset, we identified 4726 upregulated and 1570 downregulated genes in DR (Figure 3A). Intersection analysis of MR-identified proteins, aging-related genes, and differentially expressed genes revealed PXN as the sole overlapping candidate (Figure 3B), with significantly elevated expression in DR patients (p < 0.001; Figure 3C). This convergence of causal, enrichment, and transcriptomic evidence highlights PXN as a compelling molecular candidate in DR pathogenesis.

PXN as a key gene in diabetic retinopathy (DR). (A) Volcano plot of differential expression (GSE221521), showing significantly upregulated (red) and downregulated (green) genes in DR vs. controls. (B) Venn diagram integrating MR-identified DR genes, aging-related genes, and upregulated genes, revealing PXN as the only overlap. (C) Violin plot confirming PXN upregulation in DR (p < 0.001). (D) Heatmap and butterfly plot of PXN-correlated genes, highlighting co-expression patterns. (E–F) GO and KEGG enrichment of PXN-related genes, indicating pathways in extracellular matrix organization, focal adhesion, and inflammation. (G) PPI network showing PXN as a hub interacting with DR-related proteins. (H) Validation in GSE234447 (OIR mouse model) confirms PXN upregulation (p = 0.00033).
PXN is functionally linked to inflammatory signaling and matrix remodeling in DR
Correlation analysis of the GSE221521 dataset identified 604 genes significantly associated with PXN, with the top 14 visualized in heatmap and butterfly plots (Figure 3D). GO and KEGG enrichment of PXN-correlated genes implicated TNF-α signaling, T cell activation, and Th1 inflammatory responses (Figures 3E–F), aligning with established mechanisms of retinal inflammation. PPI analysis revealed strong connectivity between PXN and intracellular mediators such as TLN1 and SRC (Figure 3G), suggesting cooperative roles in cytoskeletal remodeling and leukocyte adhesion, thereby promoting endothelial dysfunction. Cross-species validation using the GSE234447 mouse oxygen-induced retinopathy (OIR) model confirmed PXN upregulation in ischemic retinas (Figure 3H), supporting its relevance in retinal vascular pathology.Collectively, multi-omic and cross-species evidence positions PXN as a central modulator of inflammation, cytoskeletal signaling, and extracellular matrix dynamics in DR, and as a promising target for therapeutic intervention.
PXN expression associates with immune cell reprogramming and regulatory signaling in DR
To explore the immunological implications of PXN in DR, we analyzed immune infiltration patterns stratified by PXN expression. CIBERSORT deconvolution revealed that high PXN expression was associated with an increased infiltration of regulatory T cells (Tregs), whereas low PXN expression correlated with higher levels of plasma cells, resting/activated CD4+ memory T cells, and γδ T cells (Figure 4A), suggesting a shift toward a regulatory-dominant immune landscape. Single-sample GSEA demonstrated that high PXN expression enriched pathways related to antigen-presenting cell co-stimulation, immune checkpoint signaling, and T cell co-inhibition (Figure 4B). Immune scoring further confirmed elevated B cells, Th1 cells, TILs, and Tregs in the high PXN group (Figure 4C). PXN expression positively correlated with M0 macrophages and Tregs (Figure 4D), suggesting coordinated modulation of innate and adaptive compartments. Despite these compositional changes, overall immune, stromal, and ESTIMATE scores remained unchanged (Figures 4E–G), indicating that PXN alters immune function without gross microenvironmental remodeling. Collectively, these findings identify PXN as an immunoregulatory modulator in DR, simultaneously enhancing Th1-driven inflammation and immune tolerance.
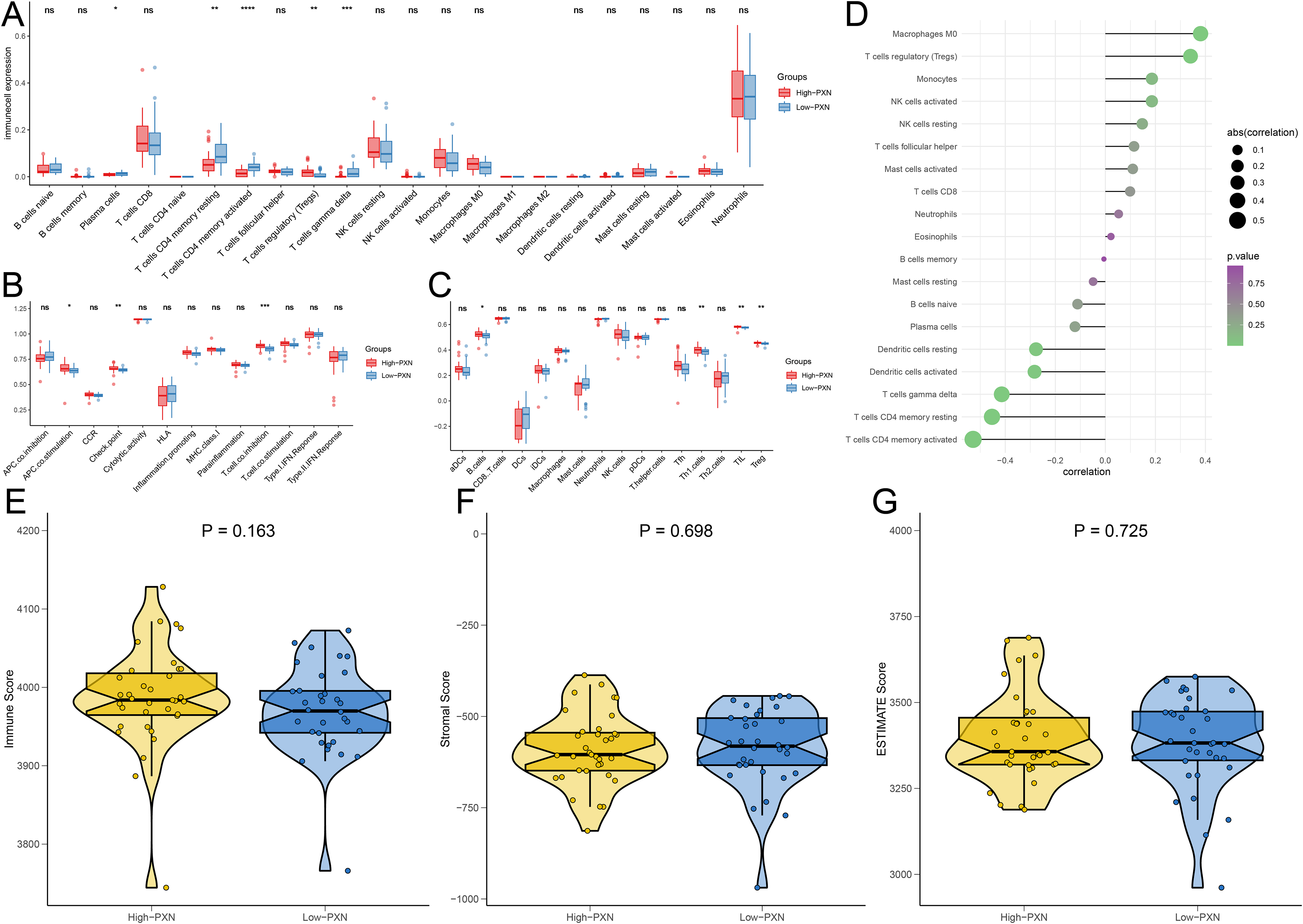
PXN and Th1-related immune processes. (A–C) Boxplots comparing infiltration levels of immune cell types and immune process activity between high and low PXN expression groups, with significant differences marked by asterisks. (D) Barplot showing correlations between PXN expression and 22 immune cell types, with bar direction and color indicating positive or negative associations. (E–G) Violin plots comparing immune, stromal, and ESTIMATE scores between PXN expression groups, showing no significant differences.
Single-cell transcriptomics identify PXN+ classical monocytes as key immunomodulators
To delineate the cellular context of PXN in DR, we analyzed single-cell RNA-seq data from the GSE248284 dataset, enabling high-resolution profiling of immune populations. UMAP clustering identified major subsets, including T cells, NK cells, and monocytes, annotated by canonical markers (Figure 5A). PXN expression was predominantly enriched in classical monocytes, with violin plots showing significant upregulation in PXN+ versus PXN− subsets (Figure 5B), indicating functional heterogeneity within the population. CellChat analysis revealed markedly enhanced interactions between PXN+ classical monocytes and proliferative MKI67+ T/NK cells compared to PXN− monocytes (Figures 5C–D). Among signaling pathways, the SELPLG–SELL axis emerged as a key mediator, with PXN+ monocytes exhibiting significantly higher ligand–receptor activity than PXN− cells (Figures 5E–H). This pathway likely facilitates immune cell recruitment and retention, thereby amplifying retinal inflammation (Figure 6). These data identify PXN+ classical monocytes as central immunoregulatory players in DR, potentially driving lymphocyte proliferation and activation via enhanced SELPLG–SELL signaling, and position PXN as a candidate therapeutic target.
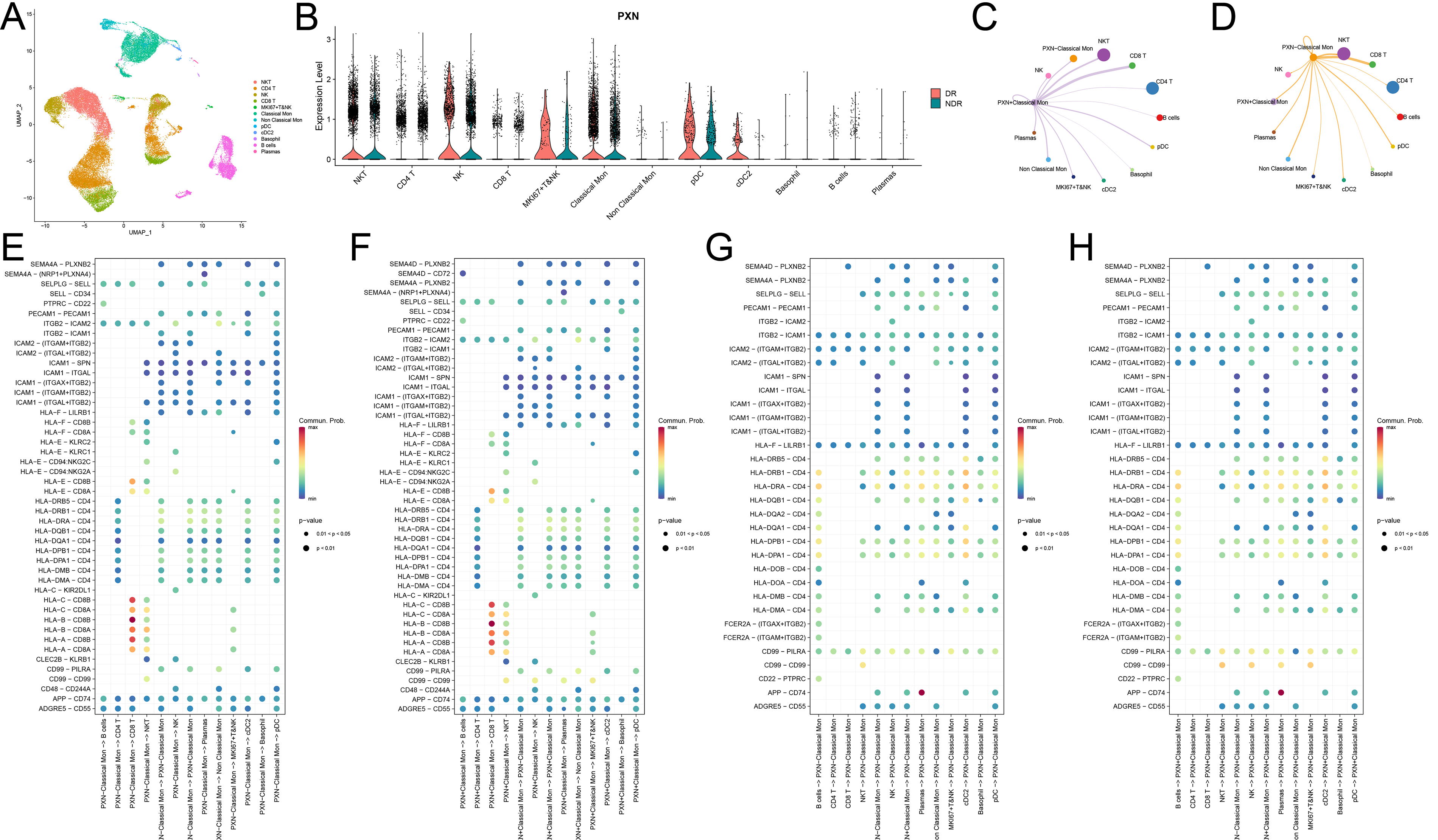
PXN+ classical monocytes enhance T and NK cell interactions. (A) UMAP plot of cell clusters from the GSE248284 single-cell RNA-Seq dataset. (B) Violin plot showing PXN predominantly upregulated in classical monocytes. (C–D) Chord diagrams illustrating stronger interactions between PXN+ classical monocytes and T/NK cells compared with PXN− cells. (E–H) Dot plots summarizing signaling pathways, with PXN+ monocytes exhibiting enriched immune activation networks as both ligands and receptors, particularly in T/NK cell communication.

PXN-mediated immune activation and inflammation in diabetic retinopathy (DR). Schematic summarizing the proposed role of PXN (paxillin) in DR pathogenesis based on Mendelian randomization and transcriptomic analyses. PXN+ monocytes, marked by SELL/SELPLG, recruit and activate MKI67+ T and NK cells, promoting proliferation and secretion of TNF-α, IFN-γ, and IL-2. These events enhance chemotaxis, Th1/M1 polarization, and cytokine-driven inflammation, contributing to retinal injury. Pathways are bioinformatically inferred and require experimental validation.
PXN regulates macrophage activation, cytokine secretion, and chemotactic gene expression in RAW264.7 cells
To validate PXN's functional role in macrophage-mediated inflammation, we performed gain- and loss-of-function assays in RAW264.7 cells. qPCR confirmed robust PXN overexpression in the OE-PXN group versus controls (p < 0.001; Figure 7A) and effective knockdown with two shRNAs (Figure 7B). CCK-8 assays showed that PXN overexpression significantly reduced macrophage viability, whereas PXN silencing promoted proliferation (both p < 0.001; Figures 7C–D). Flow cytometry demonstrated that PXN overexpression increased CD86 (M1 marker) and decreased CD206 (M2 marker) expression, indicative of pro-inflammatory polarization (Figure 7E), while knockdown reversed this phenotype (Figure 7F). ELISA confirmed that PXN overexpression elevated IL-6, IL-17, and IL-23 secretion (all p < 0.001; Figure 7G), whereas silencing reduced their levels (Figure 7H). Additionally, PXN overexpression upregulated SELL and SELPLG transcripts (p < 0.001; Figure 7I), while knockdown suppressed them (Figure 7J), implicating PXN in chemotactic regulation via the SELPLG–SELL axis. Immunofluorescence further validated that PXN overexpression increased ACE, CD86, IL-1β, IL-6, TNF-α, and CD163 protein expression, with opposite effects upon knockdown (Figure 7K). Collectively, these results demonstrate that PXN promotes macrophage M1 polarization, enhances pro-inflammatory cytokine release, and upregulates chemotactic mediators, thereby amplifying inflammatory signaling in DR.

PXN regulates macrophage activation, cytokine production, and chemotactic gene expression. (A, B) qRT-PCR analysis of PXN mRNA expression in RAW264.7 cells following PXN overexpression (OE-PXN) or knockdown (shPXN-1, shPXN-2). (C, D) CCK-8 assays showing changes in cell viability in PXN-overexpressing and PXN-silenced macrophages. (E, F) Flow cytometry detecting M1 (CD86) and M2 (CD206) markers in RAW264.7 cells upon PXN modulation. (G, H) ELISA quantification of IL-6, IL-17, and IL-23 levels in culture supernatants of PXN-overexpressing or -silenced RAW264.7 cells. (I, J) qRT-PCR detection of SELL and SELPLG mRNA expression after PXN overexpression or knockdown. (K) Representative immunofluorescence images of ACE, CD86, CD163, IL-1β, IL-6, and TNF-α expression in RAW264.7 macrophages under PXN modulation. Data are shown as mean ± SD from at least three independent experiments. Statistical significance was assessed using one-way ANOVA or Student's t-test: *p < 0.05; **p < 0.01; ***p < 0.001.
PXN regulates ACE-related oxidative stress and inflammatory responses in high glucose-stimulated macrophages
To determine whether PXN modulates RAS-mediated oxidative and inflammatory responses under diabetic-like stress, we examined ACE expression and downstream events in RAW264.7 macrophages exposed to LPS or high glucose (HG), with or without Losartan and PXN overexpression. Western blotting revealed that LPS stimulation upregulated ACE protein, an effect further enhanced by PXN overexpression, whereas PXN knockdown markedly reduced ACE expression (Figure 8A), indicating that PXN positively regulates ACE under inflammatory stress. Flow cytometry showed that HG markedly elevated intracellular ROS, which was attenuated by Losartan. However, PXN overexpression abolished this antioxidant effect, resulting in significantly increased ROS levels (Figure 8B). qPCR analysis demonstrated that HG upregulated pro-inflammatory and pro-apoptotic genes (Ace, Inos, Bax, Caspase-9) while downregulating the anti-apoptotic gene Bcl-2; Losartan reversed these changes. Notably, PXN overexpression counteracted Losartan's effects, restoring high Ace, Inos, Bax, and Caspase-9 expression and reducing Bcl-2 levels (Figure 8C). Flow cytometry of surface markers showed that HG promoted M1 polarization, whereas Losartan induced an M2-like shift. PXN overexpression reversed this shift, re-establishing a pro-inflammatory phenotype (Figure 8D). Immunofluorescence confirmed that HG increased ACE, IL-6, and CD86 expression, changes mitigated by Losartan but restored by PXN overexpression (Figure 8E). Collectively, these results indicate that PXN enhances ACE expression and amplifies oxidative and inflammatory signaling, thereby negating the protective effects of RAS blockade in HG-stimulated macrophages. PXN likely acts as an upstream modulator of RAS-driven immune activation in diabetic inflammation.

PXN promotes ACE-related oxidative stress and inflammatory polarization in high-glucose-stimulated macrophages. (A) Western blot analysis of ACE protein expression in RAW264.7 cells treated with LPS or Losartan under NC, sh-PXN, and OE-PXN conditions. (B) Flow cytometry quantification of intracellular ROS levels in RAW264.7 cells treated with HG, Losartan, and/or OE-PXN. (C) qPCR analysis of mRNA expression levels of Ace, Inos, Bax, Caspase-9, and Bcl-2 under indicated treatment conditions. (D) Flow cytometry detection of M1 (CD86) and M2 (CD206) markers in RAW264.7 macrophages subjected to HG and Losartan with or without PXN overexpression. (E) Immunofluorescence imaging of ACE, CD86, and IL-6 expression in RAW264.7 cells under the same treatments. Data are presented as mean ± SD from at least three independent replicates. Statistical comparisons were performed using one-way ANOVA or Student's t-test; *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
DR remains one of the most serious microvascular complications of DM, imposing a substantial burden on global public health systems.59,60 It is a leading cause of vision impairment and blindness in working-age adults, with prevalence rising in parallel with the global diabetes epidemic.5,61 Current treatments—laser photocoagulation, anti-VEGF therapy, and vitrectomy—offer partial benefits but are limited by the chronic, progressive nature of DR, high recurrence rates, heterogeneous patient responses, and dependence on factors such as glycemic control and disease duration.62–64 Comprehensive clinical characterization, including disease duration, glycemic status, and treatment history, is essential for interpreting immune-related findings, as chronic hyperglycemia and long-standing disease are known to modulate immune activation and influence DR progression.65,66
DR pathogenesis involves a complex interplay of metabolic dysregulation, oxidative stress, and inflammation.12,67 Hyperglycemia-induced oxidative damage to the retinal microvasculature triggers activation and infiltration of monocytes, macrophages, and T cells, driving retinal inflammation and vascular injury.10,68–70 Recognition of these immune-mediated processes has spurred interest in elucidating their molecular underpinnings to identify novel therapeutic targets.
In this study, integrated Mendelian randomization, transcriptome-wide association, and single-cell RNA sequencing analyses implicate PXN in DR progression.71–74 PXN, a cytoskeletal adaptor protein, plays a pivotal role in cell adhesion, migration, and signal transduction—processes closely linked to immune cell trafficking and vascular homeostasis.75,76 PXN upregulation in DR was associated with endothelial dysfunction, vascular leakage, and enhanced immune infiltration.77–80 Protein–protein interaction analysis positioned PXN as a central node within inflammatory and immune-related pathways, notably TNF-α signaling and Th1-mediated responses,76,81 underscoring its potential as both a biomarker and therapeutic target.
Despite these findings, PXN's candidacy as a therapeutic target requires cautious evaluation. Current evidence is largely correlative, and functional studies are needed to establish causality. Furthermore, given that PXN is integral to essential cellular processes such as adhesion, migration, and survival, systemic inhibition may lead to unintended off-target effects in organs that critically depend on cytoskeletal integrity, including the heart, liver, and kidneys. 82 Future therapeutic strategies must balance potential benefits in DR against these systemic risks, possibly through targeted delivery or pathway-specific modulation. 83
The MR analysis in this study employed single nucleotide polymorphisms (SNPs) derived from European populations in the 1000 Genomes Project. Given that our study cohort is primarily East Asian, this introduces potential population stratification bias in causal inference. Genetic architectures can differ substantially across populations, particularly for complex traits such as DR, where gene ethnicity interactions may modulate disease risk.84,85
Ethnic-specific susceptibility loci for DR have been reported,86,87 indicating that causal variants identified in European datasets may not be fully translatable to East Asian populations. While the use of large-scale European GWAS datasets ensured sufficient instrument strength for MR, this limitation underscores the need for future studies incorporating East Asian GWAS data to validate cross-ancestry generalizability and minimize bias from ancestral mismatch.
Clinical factors such as ethnicity and glycemic control may also modulate DR progression and treatment efficacy. Ethnic differences in immune response and metabolic regulation could confound genetic associations, particularly in chronic inflammatory settings. Incorporating clinical metadata would help elucidate how genetic determinants interact with patient-specific variables to shape DR progression. Although our findings nominate PXN as a promising therapeutic target, long-term suppression of PXN may have unintended effects. PXN plays a role in normal cytoskeletal remodeling and tissue repair, and its sustained inhibition might disrupt physiological processes, leading to secondary complications.82,88
The immune system is a critical driver of DR pathology, with monocyte activation and T cell infiltration playing central roles.69,89 Our data indicate that PXN+ monocytes are hyperactivated in the diabetic milieu, enhancing recruitment of T and NK cells to the retina. 90 These immune cells secrete cytokines such as IFN-γ and TNF-α, which compromise the blood–retinal barrier and contribute to macular edema.91,92 Mechanistically, PXN not only alters the chemotactic properties of monocytes but also enhances their interactions with other immune subsets. 93 In particular, PXN+ monocytes promote Th1 cell activation and proliferation, sustaining a pro-inflammatory retinal microenvironment that perpetuates vascular injury and neovascularization. 94 Notably, the magnitude of PXN+ monocyte activation and its downstream inflammatory effects may vary according to clinical parameters such as glycemic control and disease duration, further highlighting the importance of integrating genetic and clinical data in DR research.
While our understanding of PXN-mediated immune responses in DR is supported by mRNA and protein expression analyses, functional validation remains essential to establish its mechanistic role. Notably, the SELPLG–SELL axis, identified as a key pathway in PXN+ monocyte activation, was assessed only at the mRNA level via scRNA-seq. The mechanisms through which PXN regulates SELL, SELPLG, and ACE expression remain unclear. Although PXN is known to interact with transcription factors and signaling cascades, it is yet to be determined whether these effects arise from direct transcription factor binding, chromatin remodeling, or epigenetic regulation.95,96 Given PXN's role in cell adhesion and migration, a plausible hypothesis is that it modulates chromatin accessibility at the SELL and SELPLG loci in response to pro-inflammatory stimuli.17,97 Additionally, PXN's interaction with Rho GTPases may indirectly activate transcription factors such as NF-κB, which are central to immune cell recruitment and vascular leakage in DR.98,99 Experimental approaches such as chromatin immunoprecipitation (ChIP) and epigenetic profiling will be critical to elucidate these mechanisms, particularly in models that closely recapitulate the diabetic retina. The regulatory link between PXN and ACE expression also warrants further investigation. A plausible mechanism involves indirect modulation via ROS/NF-κB signaling, both of which play established roles in immune activation and inflammation in DR. 70 Hyperglycemia-induced ROS production can activate NF-κB, thereby upregulating ACE and other pro-inflammatory mediators, potentially enhancing vascular permeability and exacerbating retinal inflammation. 100 While this pathway remains speculative, targeted studies assessing PXN's influence on ROS generation and NF-κB activation in experimental DR models would provide valuable mechanistic insights.
To definitively clarify PXN+ monocyte–mediated chemotaxis in DR, future studies should employ gene knockout or knockdown strategies in experimental models. For example, CRISPR–Cas9–mediated deletion of SELPLG or SELL in myeloid cells, followed by in vivo tracking of monocyte migration during retinal inflammation, could directly assess pathway function. Alternatively, RNA interference or pharmacological inhibition of the SELPLG–SELL axis may determine whether blocking this interaction attenuates PXN-driven immune responses in DR. Enrichment analyses in this study provide compelling evidence for PXN's central role in DR. Functional annotation revealed strong associations between PXN and leukocyte-mediated immune responses, extracellular matrix degradation, and TNF-α signaling. 30 PXN-linked activation of MMPs offers a plausible mechanism for vascular instability, as MMP-mediated extracellular matrix breakdown increases vascular permeability and compromises retinal capillary integrity.101,102 These results align with prior reports of MMP involvement in DR and extend mechanistic understanding of PXN's role in exacerbating disease progression.
Single-cell RNA-seq analysis further identified PXN+ monocytes as highly interactive with T and NK cells via the SELPLG–SELL axis. This pathway promotes immune cell adhesion and migration, facilitating leukocyte infiltration into the diabetic retina. PXN expression in monocytes correlated with elevated pro-inflammatory cytokines, promoting Th1 polarization and sustaining chronic inflammation. While these findings provide a mechanistic link between systemic metabolic dysregulation and localized immune responses, they are largely based on transcriptomic correlations. Direct validation—such as co-culture assays or immunofluorescence colocalization of PXN+ monocytes with T/NK cells—remains necessary to confirm causality.Future experiments should incorporate functional validation approaches, including co-culture under diabetic-like conditions and spatial colocalization studies in retinal tissues. These would establish whether PXN directly mediates T/NK cell recruitment and activation, thereby solidifying its role in immune-driven DR pathogenesis.103,104
Functionally, our in vitro assays in RAW264.7 macrophages revealed that PXN promotes macrophage survival, induces M1 polarization, and elevates IL-6, IL-17, and IL-23 secretion—cytokines known to worsen retinal inflammation. PXN knockdown reversed these effects and reduced SELL and SELPLG expression, implicating PXN in chemotactic signaling. Immunofluorescence confirmed these changes at the protein level. However, RAW264.7 cells may not fully recapitulate human DR pathology,105,106 and validation in primary human monocytes/macrophages or retinal endothelial cells is warranted.
Under high-glucose and LPS stimulation, PXN also upregulated ACE and ROS production, linking it to oxidative stress and RAS activation.107,108 However, the pathogenesis of DR extends beyond hyperglycemia alone. 67 Incorporating these factors into future models would better reflect the in vivo retinal microenvironment. 109 Given evidence that hypoxia-inducible factors synergize with hyperglycemia to drive immune infiltration and neovascularization,9,110 PXN's role in such combined stress conditions warrants exploration.
Notably, PXN overexpression negated the anti-inflammatory and antioxidant effects of Losartan, reinstating pro-inflammatory gene expression, M1 markers, and cytokine secretion. This suggests PXN may undermine RAS blockade efficacy, highlighting the need for combination strategies targeting both PXN and RAS pathways. 111 Testing whether PXN knockdown enhances RAS inhibitor responsiveness could have direct clinical implications. 112 Correlation and immune infiltration analyses further confirmed PXN's association with increased Th1 and activated T cell infiltration, along with elevated Tregs.76,113,114 This dual pro- and anti-inflammatory modulation underscores DR's complex immune milieu and the need for targeted strategies to restore balance.
Therapeutically, PXN and its signaling partners, represent promising targets for disrupting the cycle of inflammation, immune cell recruitment, and vascular damage in DR. 115 The integration of Mendelian randomization, single-cell transcriptomics, and enrichment analyses in this study strengthens the mechanistic evidence for PXN as a driver of immune dysfunction in DR.103,116,117 By pinpointing PXN+ monocytes as key mediators of retinal injury, these findings provide a foundation for developing PXN-targeted immunomodulatory therapies. Nevertheless, our MR analysis population warrants further validation in ethnically matched cohorts. Translating these findings into clinical trials could yield new treatment strategies addressing a major unmet need in DR management.118,119
Conclusion
In summary, this study identifies PXN as a central mediator linking immune activation, inflammatory signaling, vascular dysfunction, and cellular senescence in DR. By integrating Mendelian randomization, bulk and single-cell transcriptomics, and macrophage model validation, we demonstrate that PXN promotes pro-inflammatory activation, cytokine secretion, chemotactic signaling via the SELPLG-SELL axis, and oxidative stress through ACE upregulation. These processes collectively drive immune infiltration, vascular injury, and premature retinal immune aging. Importantly, PXN overexpression attenuates the anti-inflammatory efficacy of RAS inhibition, providing mechanistic insight into therapeutic resistance. Taken together, our findings highlight PXN as both a biomarker of inflammatory and senescence-driven retinal pathology and a potential therapeutic target. Future studies should evaluate whether PXN-targeted strategies, alone or in combination with anti-VEGF or RAS blockade, can mitigate immune senescence and improve outcomes in DR.
Supplemental Material
sj-pdf-1-jra-10.1177_14703203251386803 - Supplemental material for PXN+ monocytes accelerate diabetic retinopathy progression and inflammaging via SELPLG-SELL axis-mediated immune chemotaxis and RAS modulation
Supplemental material, sj-pdf-1-jra-10.1177_14703203251386803 for PXN+ monocytes accelerate diabetic retinopathy progression and inflammaging via SELPLG-SELL axis-mediated immune chemotaxis and RAS modulation by Na Liang, Jing Li, Wenting Wang, Huifang Xu and Shaomin Peng in Journal of the Renin-Angiotensin-Aldosterone System
Footnotes
Ethics declaration
No procedures were performed on live human or animal subjects.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Scientific Research Project of the Health Commission System of Ningxia Hui Autonomous Region (Grant No. 2022-NWKY-079).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.