
Abstract
Hypothesis:
A lack of contraction of cerebral microarterioles to Ang II (“resilience”) depends on cyclooxygenase (COX) and lipocalin type prostaglandin D sythase L-PGDS producing PGD2 that activates prostaglandin D type 1 receptors (DP1Rs) and nitric oxide synthase (NOS).
Materials & Methods:
Contractions were assessed in isolated, perfused vessels and NO by fluorescence microscopy.
Results:
The mRNAs of penetrating intraparenchymal cerebral microarterioles versus renal afferent arterioles were >3000-fold greater for L-PGDS and DP1R and 5-fold for NOS III and COX 2. Larger cerebral arteries contracted with Ang II. However, cerebral microarterioles were entirely unresponsive but contracted with endothelin 1 and perfusion pressure. Ang II contractions were evoked in cerebral microarterioles from COX1 –/– mice or after blockade of COX2, L-PGDS or NOS and in deendothelialized vessels but effects of deendothelialization were lost during COX blockade. NO generation with Ang II depended on COX and also was increased by DP1R activation.
Conclusion:
The resilience of cerebral arterioles to Ang II contractions is specific for intraparenchymal microarterioles and depends on endothelial COX1 and two products that are metabolized by L-PGDS to generate PGD2 that signals via DP1Rs and NO.
Keywords
Introduction
The PO2 of the brain is highly variable and averages only 20 to 25 mmHg 1 whereas the PO2 of the outer renal cortex is quite constant averages and 42 mmHg. 2 Thus, the brain requires vascular mechanisms to limit vasoconstriction and prevent ischemia. These could be referred to as providing “resilience” to vasoconstriction. Angiotensin II (Ang II) has complex effects on cerebral blood vessels and cerebral blood flow (CBF). Thus, prolonged Ang II causes cerebrovascular oxidative stress, vasoconstriction and neurovascular dysfunction 3 that, during inhibition of nitric oxide (NO) generation, can contribute to vascular cognitive impairment. 4 However, whereas the larger cerebral and pial blood vessels constrict with Ang II when applied topically, 5 this wanes over time 6 and these vessels dilate when Ang II is perfused intraluminally. 7 Indeed, CBF does not normally change, or may even increase, during intracarotid or intravenous infusions of Ang II in mice, 8 cats, 9 piglets 7 or humans. 10 The cerebral vasodilation, or increase in CBF with Ang II depends on angiotensin type 1 receptors (AT1Rs),9,11 nitric oxide synthase (NOS) and cyclooxygenase (COX) 7 but is independent of AT2Rs and ATIVRs.9,11 However, the responsible COX-dependent prostaglandin (PG) or whether its effects are linked to NO are unknown. Direct measurement of pial artery pressure in piglets has led to the conclusion that intra-arterial Ang II decreases cerebral vascular resistance in smaller arterioles. 12 Therefore, the focus of this study is on intraparenchymal cerebral microarterioles.
Pial blood vessels give rise to penetrating intracerebral microarterioles that supply the cerebral capillaries. These microarterioles are the site of cerebrovascular coupling. 13 Occlusion of even one penetrating cerebral arteriole in rats can lead to a stroke and cognitive impairment. 14 Cerebral small vessel disease in humans can cause widespread damage with small subcortical infarcts, lacunes, cerebral microbleeds, cortical microinfarcts and white matter hyperintensities that are associated with incident stroke and dementia. 15 Thus, these small intraparenchymal blood vessels could be a focal point both for the regulation of tissue blood flow and for cerebrovascular disease. Nevertheless, because of the technical challenges, there is little direct information on their function16–18 and no reports of their direct responsiveness to Ang II.
Prostaglandin D2 (PGD2) is the major PG in cerebrospinal fluid, 19 cerebral tissue 19 and cerebral microvessels. 20 Cerebrovascular PGD2 is synthesized primarily by lipocalin-type prostaglandin D synthase (L-PGDS) 19 and activates prostaglandin D type 1 receptors (PD1Rs) 19 to cause vasodilation mediated by nitric oxide synthase (NOS) and cyclic adenosine monophosphate (cAMP). 21 Cerebral microvessels incubated with Ang II or norepinephrine release PGD2 preferentially. 22 Although PGE2 can mediate vasodilation, it constricts cerebral parenchymal arterioles. 23 Prostacyclin (PGI2) is generated by prostacyclin synthase (IPS) and has widespread vasodilator actions in blood vessels. Since these studies were designed to investigate the mechanisms of selective cerebral vs renal microarteriolar protection from vasoconstriction with Ang II, the focus was on the L-PGDS/PGD2/DP1R pathway. We tested the hypothesis that Ang II vasoconstriction of isolated, perfused cerebral microarterioles is opposed by PGD2 acting via DP1Rs and NOS. We contrasted the responses of isolated, perfused cerebral microarterioles dissected from within the frontal cortex with renal afferent arterioles (Affs) that have a robust vasoconstrictor response to Ang II. 24
Materials and methods
All procedures conformed to the Guide for Care and Use of Laboratory Animals prepared by the Institute for Laboratory Animal Research. Studies were approved by the Georgetown University Animal Care and Use Committee.
Mice
Male C57BI/6 mice (22–30 g) aged 2 to 4 months (Charles River, Inc, Germantown, MD) were fed standard mouse chow ( Na+ 0.4 g ∙ 100 g−1; Teklad Laboratory Animal Diets). 25 The knockout (–/–) COX1, TPR, superoxide dismutase 1 (SOD1) and p47phox mice have been described and validated.26–30 Knockout and wild type (+/+) mice were selected from heterozygote crossmatches and compared to littermate +/+ mice. Female mice were placed in a cage previously inhabited by a male mouse to synchronize cycling and estrous confirmed by many epithelial cells on a vaginal swab. 31
Isolated CA and Affs
Mice were euthanized under isoflurane (2% in O2). One cerebral intraparenchymal microarteriole (internal diameter 15–22 µm) was dissected from within the frontal cerebral cortex and one renal afferent arteriole (internal diameter 10–12 µm) from the outer renal cortex.
mRNA expression in CAs and Affs
Fifteen intraparenchymal CAs of six mice were pooled and 15 Affs pooled separately. They were placed in a tube with 2 mL of lysing matrix (Fisher Scientific, Santa Ana, CA 92707) containing QIAzol lysis reagent and homogenized by MP Fast Prep. Total RNA was extracted with an RNeasy Mini Kit (Qiagen Inc. Germantown, MD 20874) and analyzed by RNAseq. 32
Isolated, perfused CAs and Affs
Individual microarterioles were dissected, mounted and perfused at 40 mmHg via a pipette whose tip pressure was recorded by a calibrated intraluminal pipette.25,29,33 Perfusion at >32 mmHg generated basal tone that was assessed from the increase in wall tension and diameter in physiological solution compared to Ca2+-free solution. 33 Basal tone was apparent also in the genetic models studied.29,30,33 When stabilized, vehicle or Ang II (10−12 to 10−6 mol ∙ L−1) were added to the bath for 2 min interspersed with 4 min for recovery. Only one concentration-response curve was generated from each arteriole. This protocal does not lead to desensitization 34 and yields similar results to intraluminal Ang II. 35 Some vessels were deendothelialized by luminal perfusion with saponin (0.125 mg ∙ ml−1 15 min) and deendothelialization was confirmed by loss of relaxation with acetylcholine (10−5 mol ∙ L−1). 36 The effect of COX1 was assessed in COX1 –/– versus +/+ mice, 27 of COX2 from effects of parecoxib (10 mg∙kg−1 ip for 3 days vs vehicle; Sigma-Aldrich, St. Louis, MO 63103),37,38 of SOD1 and 3,TPR and p47phox by contrasting –/– vs +/+ mice, 24 of NOS from blockade with l-nitroarginine methyl ester (L-NAME; Sigma-Aldrich, St Louis, MO 63103; 10−4 mol∙L−1), of L-PGDS from blockade with AT56 (5 × 10–4 mol ∙ L, selective for L-PGDS vs COX and other PG synthases 39 ), of prostacyclin receptors (IPR) from blockade with RO 1138452 (10−6 mol ∙ L−1) 40 and of DP1R from activation with the stable ligand BW245c (10−6 mol ∙ L−1). 41 All drugs were added to the bath of perfused micoarterioles.
Microarteriolar nitric oxide (NO) activity with Ang II
Perfused CAs and Affs were loaded with 5 × 10−5 mol ∙ L−1 4-amino-5-methylamino -2’-7’ difluorescein diacetate (DAF-FM-DA; Sigma-Aldrich, St Louis, MO 63103). Increased fluorescence after 1 minute of Ang II (10−6 mol ∙ L−1) or BW245c (10−5 mol ∙ L−1) vs vehicle quantitated NO production. 37
Cerebral posterior communicating and basilar arteries studies
Single vessels were dissected, mounted on a myograph and Ang II contractions assessed as described in detail. 26
Statistics
Dose-response curves from groups (n = 6–10) of mice were analyzed by repeated measures 2×2 ANOVA to assess the effects of an intervention (e.g. COX inhibition) and vessel type (e.g. cerebral vs renal microarterioles) and their interaction (e.g. effects of vessel type on response to COX inhibition). Descriptive post-hoc analyses used nonparametric Wilcoxon’s statistics (GraphPad Prism, GraphPad Software) to identify the responsive arteriole. Data are presented as mean ± SEM values and p < 0.05 was considered significant.
Results
Gene expression in CAs versus Affs
Supplemental Tables S1 and S2 present selected data and approximate relative gene expression in cerebral vs renal microarterioles from the ratios of fragments per kilobase of transcripts per million mapped reads (FPKM) in the two arterioles. The genes for L-PGDS, DP1R, COX2, IPS and prostacyclin receptor (IPR) were more frequent in CAs than Affs whereas those for thromboxane A2 synthase (TxA2S), TPR and for COX1 were similar and those for prostaglandin E2 synthase (PGE2S) and prostaglandin E2 receptors were not so clearly different. While the genes for NOS I, II and III were more frequent in CAs there was little expression of AT2R or the Ang 1-7 (Mas1) receptor but rather more CA expression of ATIVR. Affs expressed AT1aR whereas CAs expressed AT1bR.
Gene expression in cultured endothelial cells (ECs) from cerebral versus renal microarterioles
Comparison of mRNA expression in endothelial cells (ECs) cultured from cerebral microarterioles vs renal microarterioles confirmed results of RNAseq that the expression of COX2, L-PGDS, DP1R and NOS III are greater in cerebral than renal microarterioles (Supplemental Figure S1) as were genes for peroxisome proliferator-activated receptor gamma (PPARℽ) and nuclear factor E2-related factor 2 (Nrf2). Incubation for 12 h with the DP1 receptor agonist BW245c increased the mRNA expression for COX2, L-PGDS, NOS III, PPARℽ and Nrf2 in ECs from cerebral, but not renal microvascular ECs.
Vascular responses of isolated, perfused CAs versus Affs
Whereas Affs had strong, dose-dependent contractions with Ang II and norepinephrine, CAs were entirely unresponsive but both contracted similarly to endothelin 1 (ET1) and perfusion pressure (myogenic responses) and relaxed similarly with acetylcholine or NONOate (Figure 1). CAs from the brain stem of mice or from the cerebral cortex of Sprague Dawley rats were similarly unresponsive to Ang II (Supplemental Figure S2) whereas both the larger basilar, and especially the posterior communicating arteries, studied on a myograph, were quite responsive to Ang II (Supplemental Figure S3). Thus, intraparenchymal CAs from the frontal cortex and brain stem and Affs have robust endothelium dependent- and independent-relaxations and robust contractions to ET1 and perfusion pressure but, in contrast to Affs or larger cerebral arteries, intraparenchymal CAs from rodents are entirely unresponsive to Ang II.

Mean ± SEM values for changes in intraluminal diameter of perfused cerebral microarterioles and renal afferent arterioles with bath additions of angiotensin II (Panel a). Norepinephrine (Panel b). Endothelin 1 (Panel c). Acetylcholine (Panel d) or NONOate (Panel e) or changes in perfusion pressure (Panel f).
Roles of Ang receptors, ROS or sex
CAs remained unresponsive to Ang II in AT1aR –/– (vs +/+) mice or after blockade of AT1Rs, AT2Rs or ATIVRs (Supplemental Figure S4) and in SOD1 or 3 –/– (vs +/+) or p47phox –/– or POLDIP2 +/– mice (Supplemental Figure S5) and in female mice (Supplemental Figure S6).
Roles of COX 1 and 2 and the endothelium
Ang II contractions became apparent in CAs from COX1 –/– (vs +/+) mice (Figure 2(a)), after COX2 blockade (Figure 2(b)) or after endothelial denudation (Figure 2(c); confirmed by loss of vasorelaxation to acetylcholine; Supplemental Figure S7). However, deendothelialization of CAs from COX-blocked mice (COX1 –/– mice with parecoxib) failed to enhance Ang II contractions (Figure 2(d)).

Mean ± SEM changes in diameter with angiotensin II added to the bath of cerebral microarterioles from cyclooxygenase 1 +/+ versus –/– mice (Panel a) or vessels from normal mice incubated with vehicle versus parecoxib (10−5 mol ∙ L−1) (Panel b), or vessels with endothelium intact versus denuded (Panel c) or vessels from cyclooxygenase 1 –/– mice with endothelium intact versus denuded (Panel d).
Contractions to Ang II (10−6 mol ∙ l−1) in COX 1 –/– (vs +/+) mice were enhanced in CAs yet reduced in Affs while COX2 blockade enhanced contractions only in CAs (Supplemental Figure 8), consistent with the absence of mRNA for COX2 in Affs (Supplemental Table S1).
Roles of TPRs and IPRs
Contractions were similar in CAs or Affs from TPR –/– versus +/+. Blockade of IPRs 40 evoked similar increases in Ang II contractions in CAs and Affs (Supplemental Figure S9).
Roles of L-PGDS, DP1Rs and NOS
Blockade of LPGDS with AT56 39 to prevent PGD2 synthesis evoked Ang II contractions in CAs that were reduced by almost 50% by activation of DP1Rs with BW245c (Figure 3(a)) while combined COX blockade (COX1 –/– + parecoxib) to prevent all PG synthesis evoked similar Ang II contractions in CAs as blockade of L-PGDS that also were reduced by about 50% by BW245c (Supplemental Figure S10A). This suggests that PGD2 is the major COX product opposing Ang II contractions in CAs and that about half of its anti-contractile effect is independent of DP1R signaling. BW245c had no effect on COX-blocked Affs (Supplemental Table S10B), consistent with the absence of mRNA for DP1R in Affs (Supplemental Table S1).
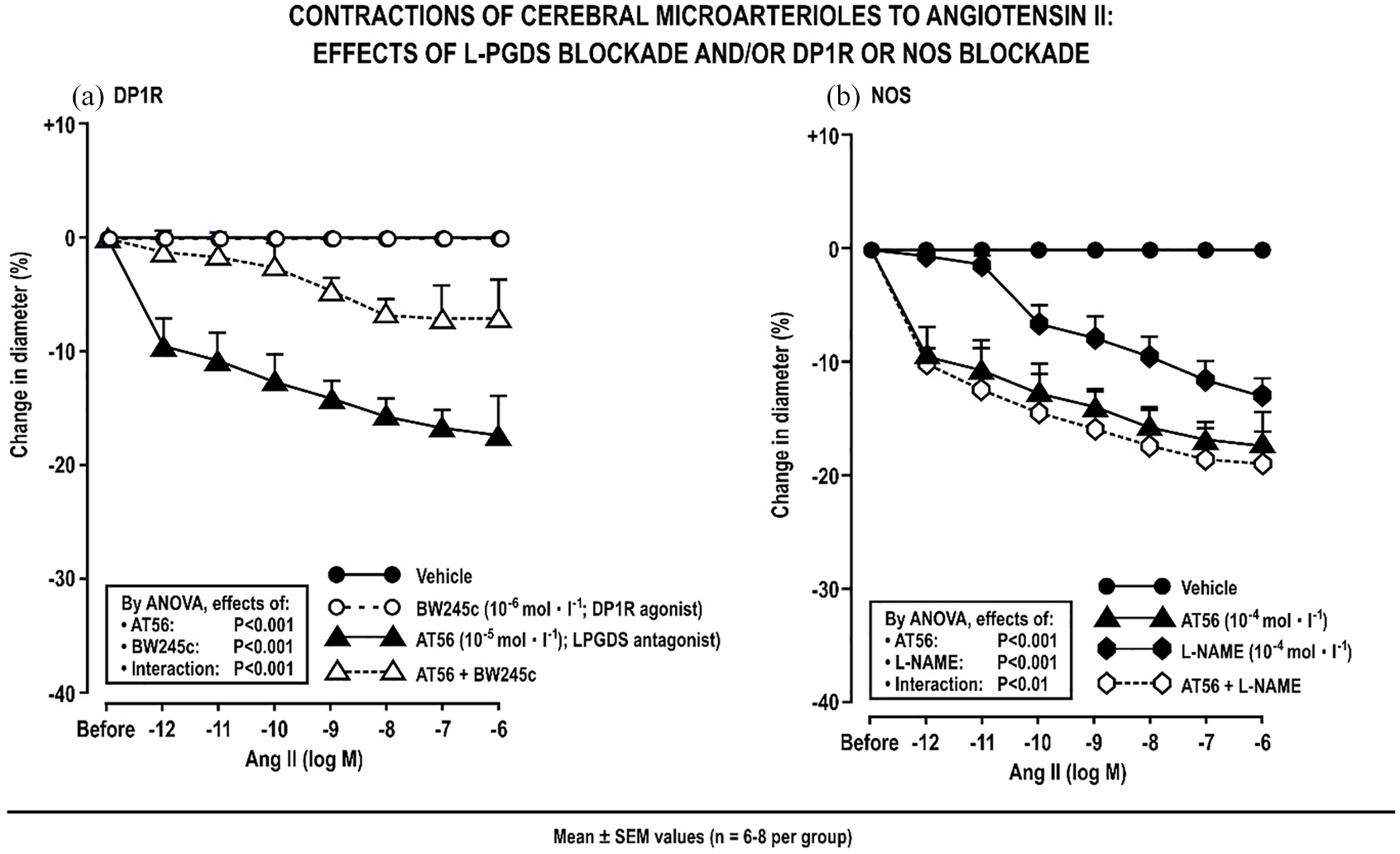
Mean ± SEM changes in diameter with angiotensin II added to the bath of cerebral microarterioles after AT56 to block lipocalin type prostaglandin D synthase or BW245c to activate prostaglandin D1 receptors (Panel a) or L-NAME to block nitric oxide synthase (Panel b).
After blockade of NOS, Ang II evoked contractions in CAs (Figure 3(b)) but AT56 enhanced Ang II contractions further in NOS-blocked CAs (Figure 3(b)) suggesting that about half of the anti-contractile effect of PGD2 was independent of NOS. However, the effects of AT56 and NOS were less than additive (significant negative interaction, Figure 3(b)) suggesting that they shared some common mechanisms. Blockade of NOS with L-NAME also enhanced Ang II contractions in Affs (Supplemental Figure S11).
Thus, endothelial COX1 + 2, LPGDS, DP1Rs and NOS provide much of the resilience of CAs to contractions with Ang II. After prevention of basal PGD2 synthesis by blockade of COX1 + 2 or LPGDS, much of the resilience of CAs to Ang II vasoconstrictions is lost but only about one half is restored by DP1R activation. Likewise, blockade of NOS also impairs the resilience of CAs to Ang II vasoconstriction but, in NOS-blocked arterioles, blockade of LPGDS enhances the resilience to Ang II vasoconstriction further by about one half. Thus, COX1 + 2 and LPGDS in CAs appear to signal predominantly via endothelial PGD2 generation that provides resilience to Ang II vasoconstriction by mechanisms that are both dependent, but also independent, of DP1R and NOS.
NO generation and action in CAs versus Affs
Increases in L-NAME-inhibitable DAF-FM-DA fluorescence with Ang II (10−6 mol ∙ L−1) were greater in CAs than Affs (Figure 4(a)) but this was almost prevented by COX1 + 2 blockade (Figure 4(b)). NO generation was also increased in CAs with BW245c (Figure 4(c)). A representative image set is shown in Supplemental Figure S12.

Mean ± SEM changes in L-NAME-inhibitable DAF-FM fluorescence of microarterioles incubated with vehicle (V) or Ang II (10−6 mol ∙ L−1) comparing cerebral microarterioles (CAs) with renal afferent arterioles (Affs) of normal mice (Panel a) or cyclooxygenase –/– mice given parecoxib (Panel b) and effects of BW245c in cerebral microarterioles (Panel c).
Discussion
The main new findings are that the genes for the vasodilator pathway of COX2/L-PGDS/DP1R/NOS are expressed robustly in CAs and in their ECs. Whereas we confirm that Affs and larger cerebral basilar and posterior communicating arteries all contract with Ang II, we find that CAs are quite unresponsive. However, both CAs and Affs contract similarly to ET 1 or perfusion pressure. This indicates that intraparenchymal CAs have a relatively selective impairment of contractions to Ang II and norepinephrine. The resilience of intraparenchymal CAs to Ang II contractions depends on COX1 and 2 and L-PGDS generating PGD2. Since both CAs and Affs share enhanced Ang II contractions after blockade of IPRs, prostacyclin cannot account for the selective resilience of CAs to Ang II contractions. Deendothelialization of CAs evokes Ang II contraction but, after deendothelialization, COX1 + 2 blockade no longer enhances Ang II contractions, thereby assigning the effects of COX to the endothelium. Ang II generates more NO in CAs than Affs and this depends on COX products while activation of DP1Rs in CAs also increases NO generation. The endothelial anti-contractile signaling of PGD2 generated by COX1 +2 and L-PGDS depends in part on DP1Rs and NOS. While the additional signaling pathways were not identified, they may entail spontaneous hydrolysis of PGD2 to 15d-PGJ2 that can activate PPARℽ 42 (Supplemental Figure S13).
Although the mRNA for AT1aR was expressed in Affs and for AT1bR in CAs, the failure of CAs to contract with Ang II cannot be ascribed to their selective AT1bR expression since they became quite responsive to Ang II after blockade of COXs, DP1Rs or NOS. Moreover, we confirm previous reports 43 that Ang II contracts small deendothelialized cerebral arterioles. The resilience to vasoconstriction is better ascribed to an offsetting anti-contractile mechanism. Mice express AT1aR in the carotid artery 44 whereas we found AT1bR expression in the intraparenchymal CAs suggesting that AT1R subtype expression varies between cerebral vessels.
Intraparenchymal CAs from the frontal cortex or brain stem of mice or from the frontal cortex of rats or from female mice were similarly unresponsive to Ang II. Thus, resilience to Ang II vasoconstriction appears to be a rather general property of intraparenchymal CAs from rodents, independent of sex. However, in confirmation of much prior work,5–7 we found larger cerebral arteries to be quite responsive to Ang II.
Although vasoconstriction with Ang II can be offset by AT2Rs, 45 mas Rs, 46 or ATIVRs, 47 their blockade failed to elicit an Ang II contraction in CAs. Moreover, contractions can be modulated by reactive oxygen species (ROS) 24 yet responses of CAs were unaffected by increasing ROS by knockout of SOD1 or 3 or decreasing ROS by knockout of p47phox or POLDIP2. Thus resilience of CAs to Ang II vasoconstriction appears independent of AT2Rs, masR, ATIVRs or ROS.
Since blockade of prostacyclin receptors enhanced contractions to Ang II in both vessels, it cannot mediate the selective resilience to Ang II vasoconstriction in CAs. The expression of the genes for PGE2 generation or action did not suggest a unique role in CAs and were not studied specifically.
PGD2 in the brain modulates sleep, pain, mood, 48 hypertension 49 and protects neurons from ischemia. 50 While L-PGDS provides resilience to Ang II contractions in CAs, restoring DP1Rs signaling with the stable DP1R agonist BW245c restores only about one half of this resilience. DP1R activation also increased NO generation. However, while NO generated in CAs also contributed to their resilience to Ang II contractions, about one half of the anti-contractile effects of L-PGDS also were independent of NO. Although the DP1R- and NOS-independent microvascular pathways were not identified, PGD2 is hydrolyzed spontaneously to 15d-PGJ2 that is a substrate for PPARℽ 42 that might mediate some effects of PGD2 in CAs. 51 Moreover, activation of DP1Rs in cerebral microarteriolar ECs upregulated PPARℽ (Supplemental Figure S1). Thus, DP1R activation may initiate interconnected sets of endothelial anti-contractile pathways that signal via DP1R, NOS and likely PPARℽ thereby providing the brain some protection from ischemia with circulating Ang II. We have reported that Ang II also activates endothelium dependent hyperpolarization 52 that can offset vasoconstriction and may have contributed to the resilience to contraction in these studies.
We acknowledge some limitations. First, RNAseq was undertaken only in one set of CAs and Affs but the results were similar in ECs cultured from these microarterioles. Second, conclusions depend on drug specificity but parecoxib or BW245c did not modify Ang II contractions of Affs consistent with their lack of mRNA for COX2 or DP1R, while RO1138452 increased Ang contractions in both vessels consistent with expression of mRNA for prostacyclin generation in both arterioles. Third, this study was confined to acute effects of Ang II on cerebral intraparenchymal microarterioles studied ex vivo, but cerebrovascular regulation by Ang II depends on intra- versus extra-vascular Ang II actions, 7 on small versus large vessels, 5 and on acute versus prolonged Ang II infusion, 3 and the superimposition of the effects of Ang II on neurovascular coupling. 3 Further work must evaluate the role of this L-PGDS pathway in the responsiveness of the cerebral circulation to Ang II. Nevertheless, after blockade generation of PGD2 or NO (Figure 3), CAs began to contract to Ang II at 10−12 to 10−10 mol ∙ L−1 that is the range of Ang II concentrations reported in plasma or cerebrospinal fluid. 53 Thus this pathway may provide some protection of the brain from vasoconstriction by circulating or brain-derived Ang II.
In conclusion, intra-parenchymal cerebral frontal microarterioles are quite resilient to Ang II contraction. This is independent of AT2, mas and ATIV receptors and of sex or ROS but depends on endothelial COX1 + 2 and L-PGDS generating PGD2 whose effects are mediated, in part, by DP1Rs and NO.
Conclusions
Cerebral small vessels contribute to stroke and vascular dementia 54 that are associated with a reduced CBF. 55 Whereas short term Ang II does not reduce CBF of normal mice, 56 prolonged infusions of Ang II into a mouse model of Alzheimer’s Disease cause AT1R-dependent cerebral vasoconstrictions and accelerated cognitive loss. 57 Augmenting the anti-contractile pathways in CAs could be beneficial in these circumstances. The reported concentrations of Ang II in plasma and cerebrospinal fluid are in the range of 10−11 to 10−9 mol ∙ l−1.58,59 These are concentrations of Ang II that contracted cerebral microarterioles after blockade of LPGDS but the contractions were prevented by BW245c (Figure 3). BW245c activates DP1Rs, is orally active in man 60 and protects the brain from hypoxic damage. 46 Remarkably, L-PGDS is the most prevalent protein in cerebrospinal fluid, is the most prevalent gene in CAs in this study (Supplemental Table S1) and can disassemble amyloid-β-fibrils. 61 Our finding that BW245c upregulates cerebral endothelial L-PGDS that mediates an anti-contractile pathway in CAs coupled with these potentially beneficial effects of L-PGDS in models of Alzheimer’s Disease suggests that DP1R activation might have a role in preventing cerebral ischemia and cognitive decline in patients at risk.
Supplemental Material
Supplement_material_9_16_2020 – Supplemental material for Endothelial prostaglandin D2 opposes angiotensin II contractions in mouse isolated perfused intracerebral microarterioles
Supplemental material, Supplement_material_9_16_2020 for Endothelial prostaglandin D2 opposes angiotensin II contractions in mouse isolated perfused intracerebral microarterioles by L Li, EY Lai, X Cao, WJ Welch and CS Wilcox in Journal of the Renin-Angiotensin-Aldosterone System
Footnotes
Acknowledgements
We acknowledge the valuable assistance of Zaiming Luo, M.D., with the molecular and cellular studies and of Robin Davisson, Ph.D., for much helpful discussion.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the NIDDK (DK-109272) and NHLBI (HL-068686), by the Walters Family Chair of Cardiovascular Research, the Georgetown University Hypertension Center, The Gildenhorn-Spiesman Family Foundation and the Smith-Kogod Family Foundation.
Supplemental material
Supplemental material for this article is available online.
Novelty and Significance
That the genes for prostaglandin D2 (PGD2) generation and response are expressed robustly in the cerebral microarterioles where they oppose vasoconstriction with angiotensin II.
That the PGD2/DP1Rsystem could provide a therapeutic window to prevent stroke and vascular dementia.
The genes for the PGD2/DP1R system are among the most plentiful in the cerebral microarterioles where they provide resilience to Ang II vasoconstriction.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.