
Abstract
Background and objective:
The fact that mineralocorticoid receptor antagonists reduce structural and functional alterations induced by cyclosporine A (CsA) indicates that aldosterone plays a key role in chronic CsA nephrotoxicity. We and other researchers have reported local renal aldosterone synthesis. To investigate local renal aldosterone’s role in chronic CsA nephrotoxicity, we evaluated the effect of eplerenone (Epl) on renal structural damage and renal dysfunction in adrenalectomized (ADX) rats, and assessed whether the therapeutic benefit was associated with reduction of transforming growth factor-β1 (TGF-β1), connective tissue growth factor (CTGF), plasminogen activator inhibitor type 1 (PAI-1) and collagen I (COL-I) expression.
Methods:
Male Sprague-Dawley rats fed a normal-sodium diet were divided in four groups: sham-ADX, ADX, CsA, or Epl. Rats in the ADX, CsA and Epl groups were adrenalectomized first. Aldosterone, sodium and potassium levels in serum and urine were measured on the second day. Two weeks later, vehicle (sham-ADX and ADX group), CsA (25mg/kg/d), or CsA and Epl (100 mg/ kg/d) combination was administrated, respectively. After six weeks, urinary protein, creatinine clearance (Ccr), tubulointerstitial fibrosis (TIF), aldosterone level in kidney, and renal aldosterone synthase CYP11B2, COL-I, TGF-β1, CTGF and PAI-1 gene expression levels were determined.
Results:
On the second day after surgery, adrenalectomized rats showed undetectable aldosterone with natriuresis, hyponatremia, decreased urinary potassium excretion and hyperpotassemia. CsA reduced Ccr, induced urinary proteins and up-regulated COL-I, TGF-β1, CTGF and PAI-1 gene expression with a significant development of TIF. Eplerenone administration prevented TIF and COL-I, TGF-β1 and PAI-1 up-regulation but did not improve renal function.
Conclusion:
Our results suggest local renal aldosterone is an important mediator of renal injury induced by CsA.
Keywords
Introduction
The introduction of cyclosporine A (CsA) has improved survival of allograft recipients and patients with autoimmune diseases; however, long-time utilization is limited in part because of chronic CsA nephrotoxicity, which is associated with progressive renal failure and striped tubulointerstitial fibrosis (TIF). Renal dysfunction appears to be the consequence of an imbalance in the release of vasoactive substances, including the activation of the renin-angiotensin-aldosterone system (RAAS), while TIF seems to be the result of several mechanisms including the increase in synthesis of some profibrotic cytokines, such as transforming growth factor-β1 (TGF-β1), connective tissue growth factor (CTGF) and plasminogen activator inhibitor type 1 (PAI-1).1–5 The up-regulation of these profibrotic cytokines results in increased synthesis as well as reduced degradation of extracellular matrix proteins, such as collagen I (COL-I).2,5,6
In recent years there has been a growing interest in the role of aldosterone in the pathophysiology of renal diseases. Clinical studies confirmed an independent effect of aldosterone in human renal disease progression despite angiotensin-converting enzyme (ACE)-inhibitor (ACEI) or angiotensin AT1 receptor blockade (ARB) treatment. 7 Aldosterone has been reported to contribute to the pathogenesis of glomerular mesangial injury, TIF and proteinuria in non-diabetic chronic kidney disease (CKD) and diabetic nephropathy.8–12 Elevated plasma aldosterone levels occur in up to half of all patients on chronic ACE inhibitor or ARB therapy, possibly due to elevated potassium levels. 13 Moreover, a contributory role of aldosterone in progression of diabetic nephropathy was shown in a diabetic rat model and in hypertensive type I diabetic patients.14,15 Antagonism of the mineralcorticoid receptor (MR, aldosterone receptor) reduces proteinuria and kidney tubulointerstitial fibrosis, independent of changes in blood pressure.16–19 In cell culture, aldosterone can activate the Rho kinase pathway, and the Rho kinase pathway is likely responsible for the profibrotic actions of aldosterone in renal proximal tubular epithelial cells via inducing epithelial-mesenchymal transition and extracellular matrix excretion. 20 MR antagonism improves proximal tubule integrity by targeting mammalian target of rapamycin (mTOR)/S6 kinase 1 (S6K1) signaling and redox status independent of changes in blood pressure. 21 In rat kidney fibroblasts aldosterone stimulates collagen synthesis through extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) signaling pathway and proliferation via activation of growth factor receptors and phosphoinositide 3-kinase (PI3K)/mitogen-activated kinase (MAPK) signaling.22,23 In mesangial cells aldosterone stimulates fibronectin synthesis through both TGF-β1-dependent and -independent pathways. 24
Although aldosterone had been believed to be synthesized only in the adrenal cortex, we and other researchers confirmed that the aldosterone synthase CYP11B2 gene, protein, and aldosterone production are locally present in the kidney, including tubular cells, mesangial cells and podocytes.25–28 Inhibition of local renal aldosterone by FAD286 (aldosterone synthase inhibitor) or spironolactone (non-selective aldosterone receptor antagonist) has been shown to decrease nuclear factor kappaB (NFκB), tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), TGF-β1, glomerular fibronectin and collagen type IV expression in diabetic nephropathy.29,30 Owing to these results, researchers concluded that local renal aldosterone contributes to development of renal inflammation, matrix formation and albuminuria, and therefore inhibition of aldosterone production in the kidney could be helpful in the management of diabetic nephropathy.29,30
Evidence from the rat model of chronic CsA nephrotoxicity has shown that aldosterone receptor blockade effectively reduced TIF and completely prevented the reduction of the glomerular filtration rate, suggesting that aldosterone contributes to renal dysfunction and structure damage.31,32 The contribution of local renal aldosterone to chronic CsA nephrotoxicity remains to be elucidated. In the present study, we hypothesized that local renal aldosterone participates in TIF of chronic CsA nephrotoxicity via enhancement of renal production of profibrotic factor, and that inhibition of local renal aldosterone, with the selective mineralocorticoid receptor antagonist eplerenone (Epl), ameliorates renal TIF.
Materials and methods
Animals
All the procedures were approved by the animal ethics committee of Qilu Hospital, Shandong University. Male Sprague-Dawley rats (Vital River Ltd, Beijing, China) weighting 300~320 g were housed in a temperature- and light-controlled environment and received a normal-sodium diet (0.284% sodium) with free access to tap water and chew. After one week for acclimatization, rats were randomized into four groups (n = 8 per group) as follows: (1) vehicle (sham-ADX); (2) ADX; (3) CsA, and (4) CsA+Epl (Epl).
Surgical procedures
Surgical procedures took place under anesthesia by sodium pentobarbital (40 mg/kg). Rats were bilaterally adrenalectomized via a dorsal approach. A small incision was made along the midline of the back just below the rib cage, connective tissue and fat were displaced, and a small hole was made through the muscle on either side of the back using blunt cut scissors. The adrenals were excised with curved forceps. 33 Each adrenal was checked that it had remained intact after excision. The second day, aldosterone, sodium and potassium in serum and urine were measured. Animals that had complete removal of both adrenals, undetectable aldosterone and electrolyte disturbance were considered completely adrenalectomized. Surgery for sham-ADX was identical, but the adrenals were not removed. All adrenalectomized rats were supplemented with dexamethasone (12 μg/kg/d, intraperitoneally) to maintain glomerular filtration rate and fasting plasma glucose. 34 Sham-ADX received sterile distilled water at a dose of 1 ml/kg/d intraperitoneally as vehicle.
Drug administration
Two weeks after surgery, sham-ADX animals received olive oil (1 ml/kg/d, subcutaneously) and sterile distilled water as the vehicle every 24 hours. Besides dexamethasone administered to the ADX, CsA and Epl groups, other drugs were given daily for four weeks as follows: The ADX group also received olive oil (1 ml/kg/d, subcutaneously) as the vehicle. The CsA group was treated with a daily dose of CsA 25 mg/kg/d administered subcutaneously. The Epl group received both 100 mg/kg/d of Epl by gastric gavage and CsA at the same dose.
Sample collection
All animals were placed in metabolic cages and urine that was spontaneously voided during every 24 hours was collected. Blood sampling was rapidly performed without anesthesia by retro-orbital sinus phlebotomy using heparinized capillary tubes, and serum was harvested immediately by centrifugation at 4°C. Urine and serum were stored at −80°C until use. At the end of the experiment, the kidneys were perfused with saline. The right kidney was removed and processed for light microscopy. The left kidney was processed for RNA analysis and aldosterone determination.
Functional studies
Urinary proteins excretion was determined using the Bradford method. Serum and urine creatinine concentration were measured with an autoanalyzer (7170A, Hitachi Co., Ltd., Tokyo, Japan) using the picric acid method. Renal creatinine clearances (Ccr) were calculated by the standard formula CCr = U · V/P, where U is the concentration in urine, V is the urine flow rate and P is plasma concentration. Aldosterone level in serum, urine and kidney were determined by enzyme immunoassay (EIA), which was performed according to the assay manufacturer’s standard protocol (Cayman Chemical Co., MI, USA). The assay has 100% specificity for aldosterone and 0.11% cross-reactivity with corticosterone. The aldosterone assay standard curve detection range is 7.8 to 500 pg/ml with a detection limit (80% B/Bo) of 14 pg/ml. Sodium and potassium levels in serum and urine were measured with an electrolyte analyzer (EA07, A&T Co., Kanagawa, Japan) using the ion selective electrode method.
Histological studies
Kidney tissues were fixed in 10% buffered formalin and then embedded in paraffin. Sections 3 μm thick were stained with Masson stain and evaluated by light microscopy by an observer masked to the treatment groups for TIF. The degree of TIF was evaluated by morphometry. For this purpose, 10 cortical periglomerular fields (magnification, ×200) were randomly selected in kidneys from the different groups. The images were recorded and the affected areas were delimited and semiquantified using the Motic Med processing and analysis system (Motic Med Imaging System Ltd., Beijing, China). Finally, the proportion of fibrosis was calculated dividing the TIF by the interstitial area.
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR)
The level of CYP11B2, COL-I, TGF-β1, CTGF and PAI-1 messenger RNA (mRNA) expression was assessed by RT-PCR. The kidney was weighed promptly and homogenized on ice, and the total renal RNA was extracted using a classic total RNA isolation kit (Sangon Co., Shanghai, China). The quality of RNA was confirmed by ethidium bromide staining in 1% agarose gel. Single-stranded cDNA was synthesized using an ImProm-II Reverse Transcription System Kit (Promega Co., Madison, WI, USA). Gene-specific primers were designed using the Gene Bank and detailed as follows: CYP11B2, forward 5’-ACGCCATCAAAGCCAACT-3’, reverse 5’-GC CACCAACAGGGTAGAG-3’; COL-I, forward 5’-CTCAGGGGCGAAGGCAACA GT-3’, reverse 5’-ATGGGCAGGCGGGAGGTCT-3’; TGF-β1, forward 5’- CGGAC TACTACGCCAAAGAAGT-3’, reverse 5’-TGGTTTTGTCATAGATTGCGTT-3’; CTGF, forward 5’-ATCCCTGCGACCCACACAAG-3’, reverse 5’-CAACTGCTT TGGAAGGACTCGC-3’; PAI-1, forward 5’-ATGAGATCAGTACTGCGGACGCC ATCTTTG-3’, reverse 5’-GCACGGAGATGGTGCTACCATCAGACTTGT-3’; glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward 5’-TGGGTGTGAACCACGAGAA-3’, reverse 5’-GGCATGG ACTGTGGTCATGA - 3’. The specificity of the primers was verified by melting curves and amplified product size using agarose gel electrophoresis. RT-PCR was performed using ABI7500 (Applied Biosystems Co., Foster City, CA, USA). Thermal cycling profile consisted of a pre-incubation step at 95°C for 60 seconds, followed by 40 cycles of denaturation (95°C, 15 seconds), annealing (58~63°C, 15 seconds) and extension (72°C, 45 seconds). Reactions were performed in triplicate, and threshold cycle numbers were averaged. Nontemplate control was used as negative control. Samples were calculated with normalization to GAPDH. Gene expression was calculated according to the formula 2(Rt–Et) × 103, where Rt is the threshold cycle number for the reference gene observed in the test sample, and Et is the threshold cycle number for the experimental gene observed in the test sample.
Statistical analysis
Data reported are mean±SEM, and all statistical analyses were calculated with SPSS 15.0. The significance of the differences between groups was tested by analysis of variance (ANOVA). Nonparametric (Spearman) correlation coefficient was calculated. The level of statistical significance was p value < 0.05.
Results
Serum and urine aldosterone before and after adrenalectomy
To determine the effect of adrenalectomy, serum and urine aldosterone levels were measured the second day after surgery. Serum and urine aldosterone levels were undetectable in the adrenalectomized groups (ADX, CsA and Epl), which indicated complete adrenal removal, and also explained the cause of hyponatremia and hyperkalemia in these groups (Figure 1(a), (b)).
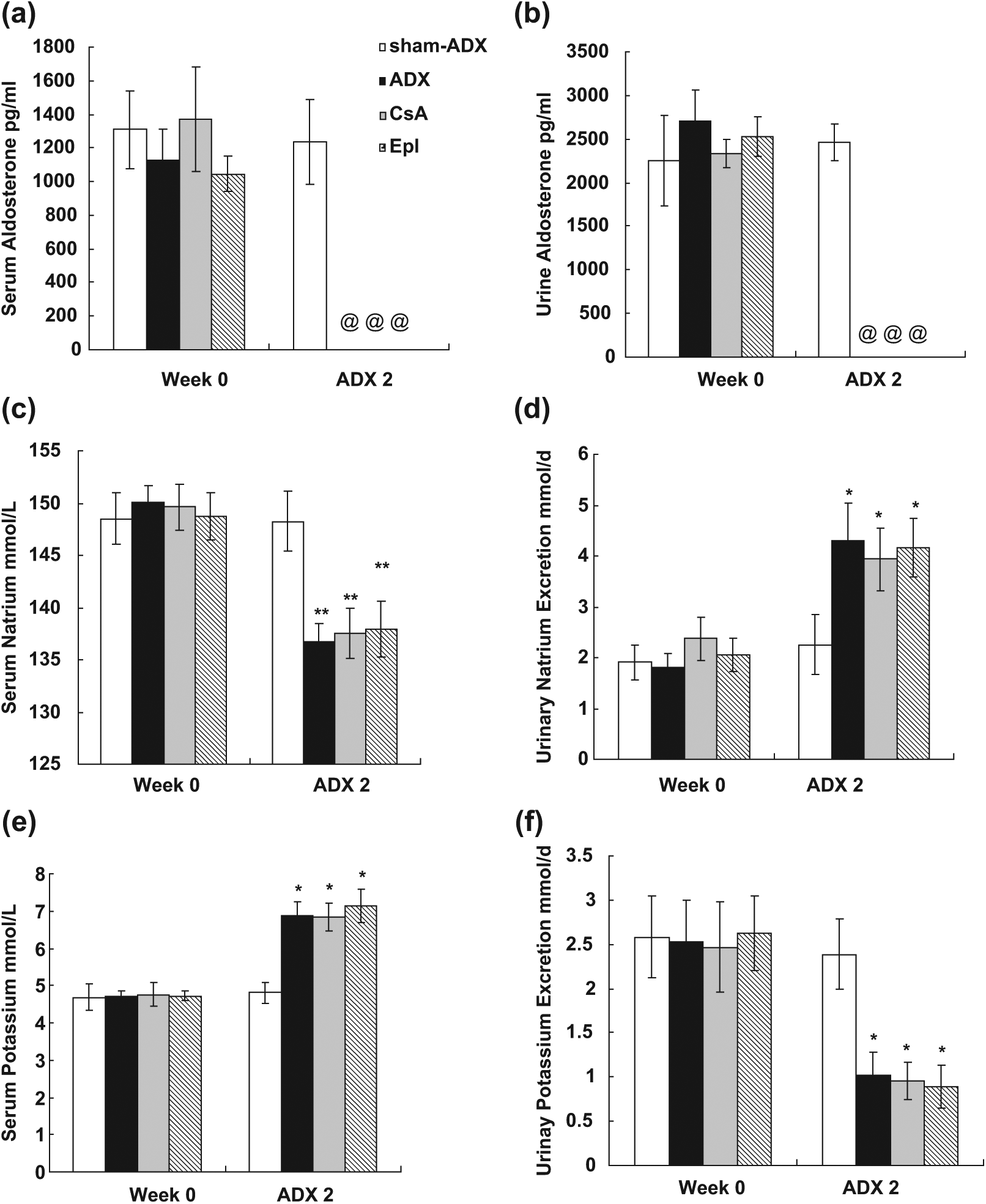
Aldosterone and electrolyte level in serum and in urinary excretion. (a) Serum aldosterone, (b) urinary aldosterone, (c) serum sodium, (d) urinary sodium excretion, (e) serum potassium, (f) urinary potassium excretion. The second day after surgery rats in the adrenalectomized groups (ADX, CsA and Epl) manifested hyponatremia and hyperkalemia accompanied by excessive urinary sodium excretion and reduced urinary potassium excretion with undetectable serum and urine aldosterone. **p < 0.01, *p < 0.05 vs. sham-ADX. @ = undetectable; ADX 2 = two days after adrenalectomy; CsA: cyclosporine A; Epl: eplerenone.
Sodium and potassium assessment
To assess the effect on electrolytes after adrenalectomy, the sodium and potassium levels in serum and urine were measured. As shown in Figure 1(c)–(f), rats in the adrenalectomized groups (ADX, CsA and Epl) manifested hyponatremia (p < 0.01, vs. sham-ADX, respectively) and hyperkalemia (p< 0.05, vs. sham-ADX, respectively) accompanied by excessive urinary sodium excretion (p < 0.05, vs. sham-ADX, respectively) and reduced urinary potassium excretion (p < 0.05, vs. sham-ADX, respectively) the second day after surgery.
Physiological and functional assessment
A mortality of 25% (two of eight rats) was observed in the ADX, CsA and Epl groups, and the animals died during the days after adrenalectomy and before drug administration. The final body weights of rats in the CsA and Epl groups were significantly lower compared with those of the sham-ADX and ADX groups (p < 0.05). Moreover, comparison between the CsA and Epl groups with the sham-ADX and ADX groups also had a significant difference (p < 0.05). Mean body weights for the CsA and Epl groups were 312±7 g and 309±9 g, respectively, at the beginning of the experiment and 271±13 g and 239±8 g, respectively, at the end. In contrast, the initial body weights for the sham-ADX and ADX groups were 313±9 g and 309±8 g, respectively, and the final body weights were 500±22 g and 356±26 g, respectively.
As Figure 2(a) shows, adrenalectomy had no effect on urinary protein excretion. Although the proteinuria levels were less or not significantly different in CsA-treated rats, as reported previously,31,35 our data indicated that CsA administration increased proteinuria levels (p < 0.01 vs. ADX), and Epl treatment resulted in a significant reduction in urinary protein excretion (p < 0.05 vs. CsA), from 11.80±2.12 mg/d in the CsA group to 5.79±2.92 mg/d in the Epl group.
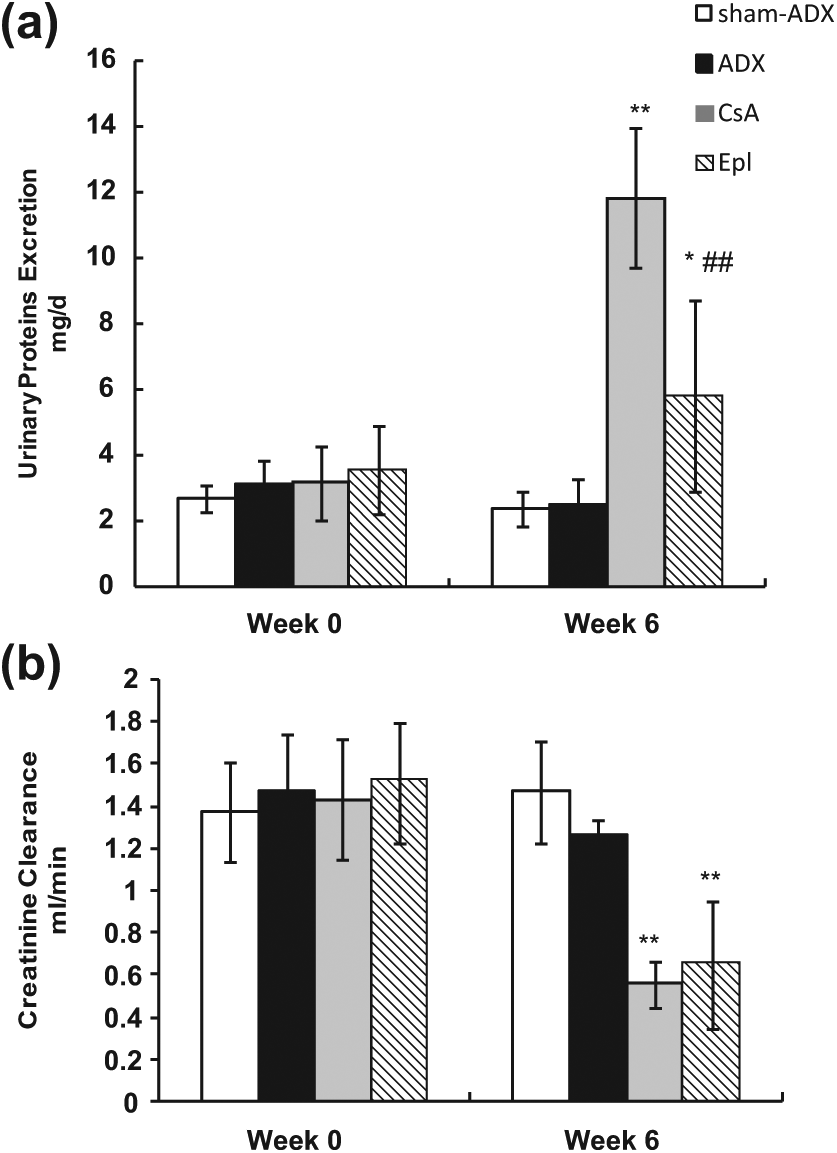
Physiological and functional assessment. (a) Urinary protein excretion and (b) creatinine clearance in the sham-ADX group, ADX group, CsA group, and Epl group. Eplerenone treatment resulted in reduction in urinary protein excretion but not in creatinine clearance. *p < 0.05, **p < 0.01 vs. ADX; ##p < 0.01 vs. CsA. ADX: adrenalectomy; CsA: cyclosporine A; Epl: eplerenone.
CCr was measured as an index of glomerular filtration rate to evaluate the effect of Epl by inhibiting local renal aldosterone. First, we confirmed the glomerular filtration rate was not influenced by adrenalectomy because the CCr value in the ADX group was not statistically different from values observed in the sham-ADX group. Second, the CsA group showed a significant decrease in CCr compared with the sham-ADX group (p < 0.01). Although the CCr value was slightly higher in rats receiving CsA and Epl than that in CsA-treated rats, the difference was not significant (Figure 2(b)).
Histological assessment
To evaluate if the new model on a normal-sodium diet is successful and Epl prevents the structural damage induced by CsA, the degree of TIF was quantified. The percentage of TIF in every section was obtained by analyzing 10 cortical periglomerular fields. Although not fed a low-sodium diet, CsA-treated rats on a normal-sodium diet after adrenalectomy had characteristic morphologic findings that were evident at the fourth week and were similar to the chronic human CsA renal lesions. Observed focal and striped TIF and tubular atrophy were most striking in the inner stripe of the outer medulla and in the medullary rays in the CsA group. A representative light microscopy image of TIF in the CsA group is shown in Figure 3(c). The concurrent administration of Epl dramatically decreased the TIF area (p < 0.05 vs. CsA, Figure 3(d)). Figure 3(a) and (b) depict that adrenalectomy did not induce structural injury. These results suggest that at least part of the CsA-induced TIF is mediated by local renal aldosterone.
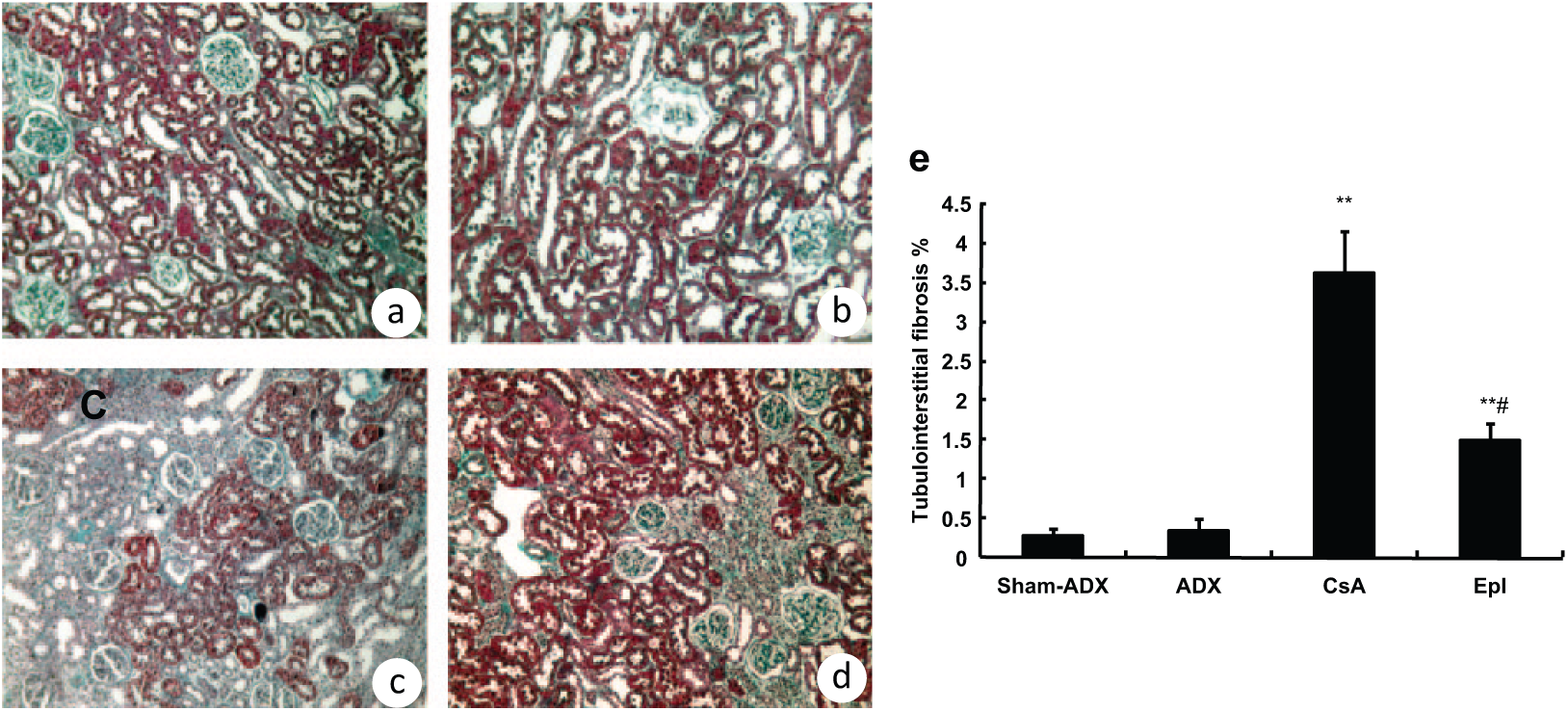
Effect of local renal aldosterone blockade with Epl on tubulointerstitial fibrosis. (a) Sham-ADX rat kidney. (b) ADX. No visible tubulointerstitial or tubular atrophy is observed. (c) The ADX animal treated with CsA. There is a diffuse tubulointerstitial fibrosis and tubular atrophy. (d) CsA and Epl-treated rat kidney after ADX. Note the decrease in interstitial fibrosis and tubular atrophy. Masson stain, magnification ×200. (e) Comparison of mean percentage of tubulointerstitial fibrosis areas. There was a significant increase in tubulointerstitial percentage in CsA-treated rats and a dramatic decrease in CsA and Epl combination treated rats. There was no significant difference between sham-ADX and ADX. Mean percentage of tubulointerstitial fibrosis area in every section obtained in 10 fields. **p < 0.01 vs. sham-ADX; # p < 0.05 vs. CsA. ADX: adrenalectomy; CsA: cyclosporine A; Epl: eplerenone.
Molecular studies
COL-I, TGF-β1, CTGF and PAI-1 mRNA expression were evaluated to understand the mechanisms by which local renal aldosterone blockade benefit the kidney from CsA toxicity. The mRNA values in these parameters in the ADX group were not statistically different from values observed in the sham-ADX group. COL-I, TGF-β1, CTGF and PAI-1 mRNA relative expression levels in CsA-treated animals had notable increases (p < 0.05, respectively), which were 34.41±1.71, 17.41±2.63, 16.15±5.10 and 1.37±0.21, respectively, compared with values in the sham-ADX group, which were 15.79±4.41, 7.58±0.80, 8.22±0.91 and 0.40±0.09, respectively. In contrast, in the Epl group relative values of these parameters were 25.88±2.68, 10.17±1.95, 14.56±1.55 and 0.33±0.13, respectively. Concomitant administration of Epl significantly decreased the mRNA expression of COL-I, TGF-β1 and PAI-1 (p < 0.05 vs. CsA, respectively), and though a slight decrease in CTGF expression was observed there was no significant difference as compared with the CsA group (Figure 4).
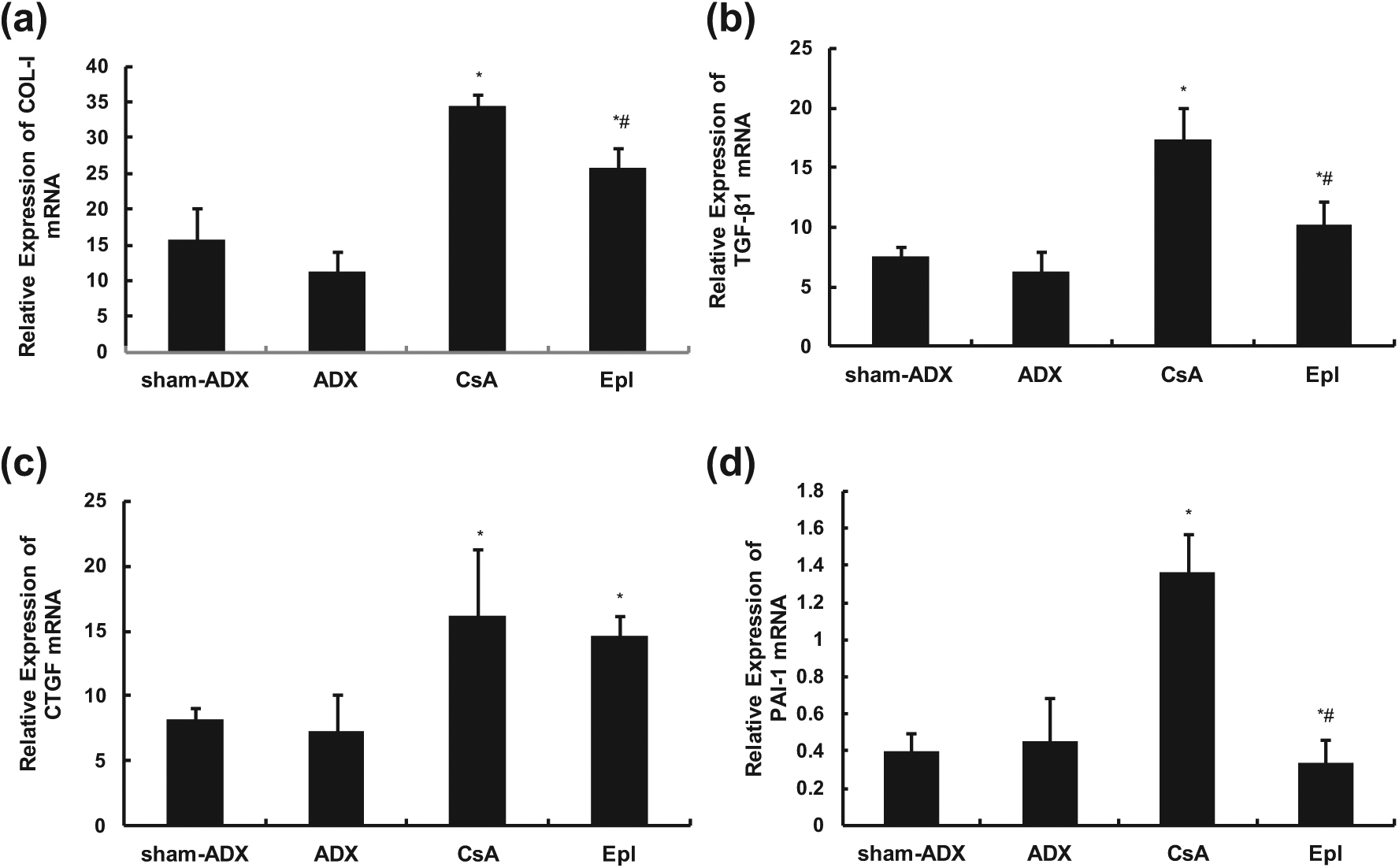
Relative expression of renal COL-I (a), TGF-β1 (b), CTGF (c) and PAI-1 (d) mRNA (real-time PCR). Concomitant administration of eplerenone significantly decreased the mRNA expression of COL-I, TGF-β1 and PAI-1, and though a slight decrease in CTGF expression was observed, there was no significant difference as compared with the CsA group. *p < 0.05 vs. sham-ADX; # p < 0.05 vs. CsA. ADX: adrenalectomy; CsA: cyclosporine A; Epl: eplerenone; COL-I: collagen I; TGF-β1: transforming growth factor-β1; CTGF: connective tissue growth factor; PAI-1: plasminogen activator inhibitor type 1; mRNA: messenger RNA: PCR: polymerase chain reaction.
Renal aldosterone and its synthase assay
Renal aldosterone synthase CYP11B2 mRNA expression and local renal aldosterone levels in adrenalectomized rats were higher than that in the sham-ADX group (p < 0.05 and p < 0.01, respectively). Although CYP11B2 gene expression showed no significant difference between the ADX group and the CsA group, the local renal aldosterone level in the CsA group was elevated compared to that in the ADX group (p < 0.01). This result suggested that CsA might stimulate the secretion of local renal aldosterone but not through the pathway of CYP11B2 gene expression. However, the underlying molecular mechanism needs further research. Renal aldosterone synthase mRNA and local renal aldosterone levels were higher in rats that received CsA and Epl combined compared with values in rats administrated CsA alone (p < 0.05 and p < 0.01, respectively). We consider it a compensatory response to inhibition of local renal aldosterone. These data are shown in Figure 5.

Renal local aldosterone levels (a) and aldosterone synthase CYP11B2 mRNA relative expression (b). Adrenalectomy elevated CYP11B2 mRNA expression and local renal aldosterone level. Local renal aldosterone level in the CsA group was higher than that in the ADX group. Both gene and protein expression was elevated in rats that received CsA and Epl combined compared with values in rats administered CsA alone. **p < 0.01, *p < 0.05 vs. sham-ADX; $ p < 0.01 vs. ADX; # p < 0.05 vs. CsA. ADX 2: two days after adrenalectomy; CsA: cyclosporine A; Epl: eplerenone; mRNA: messenger RNA.
Discussion
In this research, for the first time we developed a novel rat model of chronic CsA nephrotoxicity with a normal-sodium diet and adrenalectomy, which distinguishes it from the previously reported model with a low-salt diet.36,37 Silva et al. 38 have previously established a rat model of chronic CsA-stimulated renal injury with a normal-sodium diet. In this model, they reported the decreased glomerular filtration after CsA stimulation. However, they did not present the exact differences of renal microscopic pathological characteristics compared with the classic chronic renal injury model using a low-salt diet. In our research, rats receiving CsA and a normal-salt diet for 28 days after adrenalectomy showed a decreased Ccr and focal TIF in a striped pattern, and moreover, focal and striped TIF were most severe in the inner stripe of the outer medulla and in the medullary rays, which mimic effects of a low-salt diet. 36 Meanwhile, rats receiving adrenalectomy alone did not show renal functional or structural changes. Sodium depletion is considered critical for the development of structural changes in chronic nephrotoxicity induced by CsA.36,37 Other than sodium depletion that was achieved by a low-sodium diet in the past, we learned this by adrenalectomy, which was performed to observe local renal aldosterone. Although sodium balance was observed in adrenalectomized rats (ADX and CsA group) at the endpoint of the experiment (data not shown), we presume transitory sodium depletion induced by adrenalectomy had emerged in this model because serum aldosterone was dismissed, which resulted in hyponatremia the second day after adrenalectomy.
The mechanism whereby sodium depletion accelerates the chronic injury is unclear. One reasonable possibility would be through activation of the RAAS, which could up-regulate profibrotic factors, such as TGF-β1, CTGF, PAI-1, that contribute to renal fibrotic processes by increasing the production and decreasing the degradation of extracellular matrix proteins.3,5,39,40
After circulating aldosterone was eliminated by adrenalectomy, the role of local renal aldosterone was illuminated independently. In this research, we observed the effect of Epl, a selective aldosterone receptor antagonist, by inhibition of local renal aldosterone in chronic CsA nephrotoxicity. Epl administration reduced COL-І, TGF-β1 and PAI-1 expression, and consequently prevented TIF in this model.
It is well recognized that TGF-β1 plays an important role in CsA-induced TIF, by stimulating accumulation of extracellular matrix proteins.41–43 Instead of mediation by angiotensin II (Ang II), recent reports reveal that CsA-induced overexpression of TGF-β1 is mediated by aldosterone, since its effects can be prevented by mineralocorticoid receptor antagonist. 28 With our data, we propose that this effect is in part induced by local renal aldosterone because up-regulation of TGF-β1 is mitigated after blocking local renal aldosterone by Epl.
Although CTGF is generally considered a pro-fibrotic growth factor that acts downstream of TGF-β1, 43 whose promoter contains a unique TGF-β1 response element and expression potently induced by TGF-β1, 44 the relationship between TGF-β1 and CTGF is debated in chronic CsA nephrotoxicity. McMorrow et al. 45 showed CTGF serves as downstream mediator of TGF-β1 pro-fibrotic activity on CsA-induced epithelial-mesenchymal transition in human renal proximal tubular epithelial cells. Shihab et al. 40 observed TGF-β1 may be exerting its pro-fibrotic actions in a manner independent of CTGF in chronic CsA nephrotoxicity on a low-salt diet. In this research, Epl inhibited up-regulation of TGF-β1 mRNA expression induced by CsA but this was not accompanied by CTGF down-regulation, which indicates that local renal aldosterone enforces a pro-fibrotic effect by the TGF-β1 pathway but in a manner independent of CTGF.
PAI-1 is the major inhibitor of tissue-type plasminogen activator in vivo and thus contributes to extracellular matrix deposition. 46 Okada et al. 5 reported angiotensin-converting enzyme blockade (ACEI) can decrease PAI-1 expression, thereby facilitating matrix degradation to reduce TIF in chronic CsA nephrotoxicity. Their research did not demonstrate down-regulation of PAI-1 induced by Ang II or aldosterone, since ACEI also partly reduces aldosterone production. Our current research shows local renal aldosterone induces PAI-1 up-regulation because it is inhibited by Epl.
Theoretically Epl inhibits local renal aldosterone; persistent sodium depletion in rats administered CsA and Epl combined would result in persistent RAAS activation and more severe structure injury than rats that received CsA alone. Interestingly, however, Epl reduced TIF in our research. As our findings indicated, the possible mechanism is Epl abrogated up-regulation of TGF-β1 and PAI-1 expression by inhibiting local renal aldosterone. This demonstrates that at least in part the fibrotic effect in chronic nephrotoxicity is induced by local renal aldosterone and associated with TGF-β1 and PAI-1 overexpression.
Functional abnormality induced by CsA is associated with imbalance in the release of vasoactive substance that results in reduction of renal plasma flow and ultrafiltration coefficient, including an increase in vasoconstrictor factors as well as reduction of vasodilator factors.46–51 RAAS would be significantly activated by persistent sodium depletion in an Epl group, and in turn, both Ang II and aldosterone production would be multiplied. Ang II is an important vasoconstrictor factor in chronic CsA nephrotoxicity, and either ACEI or angiotensin AT1 receptor blockade (ARB) can ameliorate structural and functional damage.1,41,52,53 Despite reduced structural damage, inhibition of local renal aldosterone did not prevent renal dysfunction induced by CsA in our research. In this regard, we hypothesize that in this model local renal aldosterone may contribute more to structural damage, and Ang II to functional damage.
Another possible reason why we did not observe an improvement of renal function might be the concentration of Epl. If the administered dose of Epl was set as 100 mg/kg/d, the plasma level of Epl was usually less than 100 nM, which is significantly lower than the IC50 value of Epl (360 mM).54,55 In this present study, the Epl receptors might not have been fully blocked and the protective ability of Epl for renal function not completely activated. It seems that we should administrate a higher dose of Epl to undertake the full function of receptor blockade and renal protection. However, the possible side effects of Epl on blood potassium must be taken into consideration. Therefore, the appropriate administered dose of Epl must be set with maximum organ protection and minimum side effects of potassium in future research.
Circulating aldosterone is principally secreted in the glomerulosa zone of the adrenal cortex and usually results in the conversion of cholesterol to aldosterone. The level of circulating aldosterone secretion is mainly attributed to the regulation of Ang II and potassium. In addition, adrenocorticotropic hormone (ACTH), neural mediators and natriuretic factors can also affect the production of circulating aldosterone. It seems that circulating aldosterone has a few similarities with local aldosterone, for example, the similar regulators (potassium and Ang II) and the similar effect on organ fibrosis/sclerosis. However, there is a huge difference between the two of them. Circulating aldosterone plays an important role in fluid and electrolyte balance, whereas local aldosterone acts on target cells in an autocrine or paracrine manner. Of great interest, we demonstrated that the concentration of local renal aldosterone was 1000 times that in the heart (Figure 5(a) 10000 pg/g vs.10 pg/g). Luik et al. 56 reported the increased local aldosterone level but a low circulating aldosterone in the model of diabetes. Siragy and Xue 29 also demonstrated the detectable local renal aldosterone and CYP11B2 gene expression and undetectable circulating plasma aldosterone concentration 14 weeks after postadrenalectomy. These phenomena might suggest that the local renal aldosterone could not be directly reflected by circulating plasma aldosterone. Furthermore, considering the fact that the half-life of circulating aldosterone is less than 30 minutes, it is highly unlikely that local renal aldosterone is picked up from the circulation. Therefore, we did not perform the determination of circulating aldosterone in this present study.
In conclusion, we have demonstrated here the renoprotective effects of Epl in a novel rat model of chronic CsA nephrotoxicity, which includes attenuation of proteinuria and inhibition of TIF. Such beneficial effects are associated with the inhibition of local renal aldosterone. Inhibition of local renal aldosterone synthase is a potential therapeutic tool in the management of chronic CsA nephrotoxicity via reducing proteinuria and TIF.
Footnotes
Acknowledgements
The authors would like to thank Hong-Rui Dong for technical assistance.
Conflict of interest
None declared.
Funding
This work was supported by the National Natural Science Foundation of China (no. 30500246).