
Abstract
Introduction:
The nuclear factor-κB (NF-κB) is an important regulator of the inflammatory response. Angiotensin II (Ang II) activates the NF-κB pathway linked to renal inflammation. Although both AT1 and AT2 receptors are involved in Ang II-mediated NF-κB activation, the biological processes mediated by each receptor are not fully characterized. Interleukin-1β (IL-1β) is an important macrophage-derived cytokine that regulates immune and inflammatory processes, activating intracellular pathways shared with Ang II, including the NF-κB.
Materials and methods:
In vitro studies were done in primary cultured rat mesangial cells. NF-κB pathway was evaluated by phosphorylated levels of p65/IκB and DNA binding activity. The Ang II receptor subtype was determined by pretreatment with AT1 and AT2 antagonists.
Results:
In mesangial cells the simultaneous presence of Ang II and IL-1β caused a synergistic activation of the NF-κB pathway and a marked upregulation of proinflammatory factors under NF-κB control, including monocyte chemoattractant protein-1. The AT1, but not AT2, antagonist abolished the synergistic effect on NF-κB activation and proinflammatory genes caused by coincubation of Ang II and IL-1β.
Conclusions:
These data indicates that Ang II, via AT1/NF-κB pathway activation, cooperates with IL-β to increase the inflammatory response in mesangial cells.
Introduction
The mechanisms of progressive glomerulonephritis remain mostly unknown. Many studies have found renal activation of the renin-angiotensin system (RAS) and increased local angiotensin II (Ang II) production in human and experimental kidney diseases.1,2 Blockade of Ang II by using angiotensin-converting enzyme (ACE) inhibitors or Ang receptor type 1 (AT1) antagonists, are one of the best options to treat human kidney diseases. 1 Ang II blockers reduce proteinuria, delay renal fibrosis and preserve kidney function. However, use of Ang II blockers does not prevent disease progression and therefore it is necessary to investigate further the mechanisms of Ang II renal damage.
Experimental studies have shown that Ang II actively participates in the regulation of the inflammatory response in the kidney, being the activation of the nuclear factor-κB (NF-κB) pathway an important mechanism of Ang II–induced inflammatory process.2,3 Ang II binds to specific receptors, AT1 and AT2, to activate cellular responses.3,4 The AT1 receptor mediates vasoconstriction, upregulation of growth factors and fibrosis.3,4 In several cell types, Ang II activates NF-κB via AT2 receptors. 5 Interestingly, both AT1 and AT2 receptors are involved in Ang II-mediated NF-κB activation in the kidney. 6 However, the biological processes mediated by each receptor are not fully characterised.
During renal injury, many growth factors and cytokines are upregulated, and it is possible that those factors may influence the response of renal cells to Ang II. 2 The macrophage derived cytokine interleukin-1β (IL-1β) exerts a broad range of biological activities. 7 IL-1β is a proinflammatory cytokine that participates in human and experimental renal diseases.8–11 Initial studies in the model of anti-Thy-1 nephritis demonstrated that IL-1β receptor blockade reduced glomerular macrophage accumulation. 8 More recently in an experimental model of type II diabetes the IL-1β receptor antagonist, anakinra, presented protective effects, 12 showing the involvement of this cytokine in renal inflammation. IL-1β activates several intracellular pathways shared with Ang II, including the NF-κB pathway.2,7,13–15 We describe here how the simultaneous presence of Ang II and the proinflammatory cytokine IL-1β in rat mesangial cells amplified the inflammatory response, characterised by a synergism of NF-κB activation and upregulation of proinflammatory mediators, including monocyte chemoattractant protein-1 (MCP-1) and interleukin-6 (IL-6), by a process mediated by AT1. These data indicate that in inflammatory conditions, the AT1 blockade could exert additional anti-inflammatory properties by modulating cytokine-induced NF-κB activation.
Materials and methods
Reagents
Recombinant human IL-1β was obtained from Peprotech. Ang II was obtained from Tocris. NF-κB consensus oligonucleotide was from Promega Corp. The AT1 and AT2 receptor antagonist used were Valsartan (Novartis) and PD123319 (Sigma). The inhibitors used were from: BAY11-7082 and Parthenolide (NF-κB inhibitors) from Calbiochem. Cells were preincubated with all inhibitors for one hour before adding the stimulus. None of the inhibitors were toxic at the doses used. Culture reagents were purchased from Gibco BRL.
Cell culture studies
Rat mesangial cells (MCs) were cultured from isolated glomeruli by several sieving techniques and differential centrifugations.16,17 Glomerular mesangial cells were characterised by phase contrast microscopy, presenting positive staining for desmin and vimentin, and negative staining for keratin and factor VIII antigen, that exclude epithelial and endothelial contamination, respectively. Cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium, supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine in the presence of 20% heat-inactivated foetal bovine serum (FBS) and cultured at 37ºC in 5% CO2 atmosphere. At confluence cells were made quiescent for 48 h in RPMI with 0.5% FBS medium then different studies were performed. The experiments were done with cultured MCs with 0 or 1 passage.
Gene studies
RNA was isolated by Tripure (Roche, Barcelona, Spain). cDNA was synthesised using High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, California, USA) using 2 µg of total RNA primed with ramdon hexamer primers following the manufacturer’s instructions. Real-time polymerase chain reaction (PCR) reactions were performed on ABI Prism 7500 sequence detection PCR system (Applied Biosystems) according to manufacturer’s protocol. Assay IDs used were: IL-6, Rn00561420_m1; MCP-1, Rn00580555_m1; Vascular Cell Adhesion Molecule 1 (VCAM-1), Rn00563627_m1; IL-1β, Rn00580432_m1. Data were normalised with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and 18S ribosomal RNA expression (assay IDs: Rn99999916_s1 and Hs99999901_m1 respectively). The mRNA copy numbers were calculated for each sample by the instrument software using Ct value (‘arithmetic fit point analysis for the lightcycler’). Results were expressed in copy numbers, calculated relative to unstimulated cells, after normalisation against GAPDH and 18S.
Protein studies
Cells were homogenised in lysis buffer (170 mM Tris HCl, 22% glycerol, 2.2% sodium dodecyl sulphate (SDS), 0.1 mM phenylmethylsulphonyl fluoride, ortovanadate and protease inhibitor cocktail) and then separated by SDS-polyacrilamide gel electrophoresis. IκBα and phosphorylated-IκBα (p-IκBα) protein levels were determined in cytosolic protein extracts by Western blot analysis. 17 Proteins were quantified in all samples by bicinchoninic acid assay (BCA) method and a fixed amount of protein (50 µg) was loaded in each lane. The quality of proteins and efficacy of protein transfer was evaluated by Ponceau red staining. α-Tubulin or GAPDH were used as loading control. Antibodies employed were: phospho-p65 (Ser536) (#3031) from Cell Signaling, phospho-IκB Kinase α (IKKα) (Thr23) (sc-101706), total IκBα (sc-371), phospho-IκBα (Ser32) (sc-8404), total IκBα (sc-371), and IL1β receptor antibodies were from Santa Cruz Biotechnology, GAPDH (MAB374) was from Chemicon International, α-Tubulin (T5168) was from Sigma and peroxidase-conjugated secondary antibodies (Amersham). MCP-1 release was evaluated by enzyme-linked immunosorbent assay (ELISA) (RayBiotech).
Immunofluorescence
MCs were seeded in 24-well plates over crystal coverslips. After a 24 h serum-starvation step, cells were stimulated for indicated times. Then, cells were fixed in Merckofix (Merck, Millipore, Temecula, California, USA), treated with 0.1% Triton-X100, blocked with 4% BSA in PBS and incubated with primary antibody against p65 subunit (sc-372, Santa Cruz Biotechnology) overnight at 4ºC. After washing, cells were incubated with fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Sigma) and nuclei were stained with 1 µg/mL propidium iodide. The absence of primary antibody was the negative control. Samples were mounted in Mowiol 40-88 (Sigma) and examined by a laser scanning confocal microscope (Leica TSC SP5).
Electrophoretic mobility shift assay (EMSA)
Nuclear and cytosolic extracts were prepared by homogenisation and centrifugation as previously described. 16 NF-κB DNA binding activity was determined in nuclear extracts by binding with labelled NF-κB consensus and electrophoresis. The NF-κB consensus oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGGC-3′) was 32 P-end- labelled by incubation for 10 min at 37ºC with 10 U of T4 polynucleotide kinase (Promega) in a reaction containing 10 µci of [γ- 32 P]ATP, 70 mM Tris-HCl, 10 mM MgCl2, and 5 mM Dithiothreitol (DTT). The reaction was stopped by the addition of Ethylenediaminetetraacetic acid (EDTA) to a final concentration of 0.05 M. Nuclear or cellular protein (10 µg) was equilibrated for 10 min in a binding buffer containing 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), and 50 µl/ml of poly(dI-dC) (Pharmacia, Uppsala, Sweden). NF-κB activity was quantified in 10 µg of total protein of each pool, and experiments were done in duplicate. To assess the specificity of the reaction, the following controls were done: negative assay without cellular extracts; and competition assays with a 100-fold excess of unlabelled NF-κB, mutant NF-κB, an unspecific oligonucleotides. When competition assays were done, the unlabelled probe was added to this buffer 10 min before the addition of the labelled probe. The labelled probe (0.35 pmol) was added to the reaction and incubated for 20 min at room temperature. The reaction was stopped by adding gel loading buffer (250 mM Tri-HCl, 0.2% bromophenol blue, 0.2% xylene cyanol, and 40% glycerol), and run on a nondenaturing, 4% acrylamide gel at 100 V at room temperature in 0.25% Tris-Borate-EDTA buffer (TBE) . The gel was dried and exposed to X-ray film.
Statistical analysis
The autoradiographs were scanned using the GS-800 Calibrated Densitometer (Quantity One software from Bio-Rad). Results are expressed as n-fold over control as mean±standard error of the mean (SEM) of experiments made. One-way analysis of variance (ANOVA) was used to compare gene and/or protein expression levels between groups. When statistical significance was found, the Bonferroni post-hoc comparison test was used to identify differences between groups. Differences were considered significant at p<0.05. Statistical analyses were performed using the SPSS statistical software, version 16.0.
Results
The simultaneous presence of Ang II and IL-1β induces a synergistic effect on NF-κB pathway activation in cultured mesangial cells
In resting cells, NF-κB remains in the cytosol as an inactive complex, formed by the p50 and p65 subunits bound to the inhibitory subunit IκBα. 18 One of the earliest events in the canonical NF-κB pathway is the activation of the IKK complex, 19 followed by the phosphorylation of the components of NF-κB complex IκBα and p65 subunit,18,20 therefore phosphorylation levels of these proteins were used as markers of NF-κB activation. In cultured rat mesangial cells, both Ang II and IL-1β induced the phosphorylation of IKKα, IκBα and p65, alone or in coincubation (Figure 1). The phosphorylation of the inhibitory IκBα subunit targets it for an ubiquitin-proteasome dependent degradation. 18 Both Ang II and IL-1β induced the diminution of cytosolic IκBα protein levels, indicating that this protein is degraded (Figure 1(c) and 1(d)). Interestingly, cells co-incubated with both factors, these effects were more marked, as clearly shown by the ratio of phosphorylated-IκBα versus total IκBα (Figure 1(c) and 1(d)).
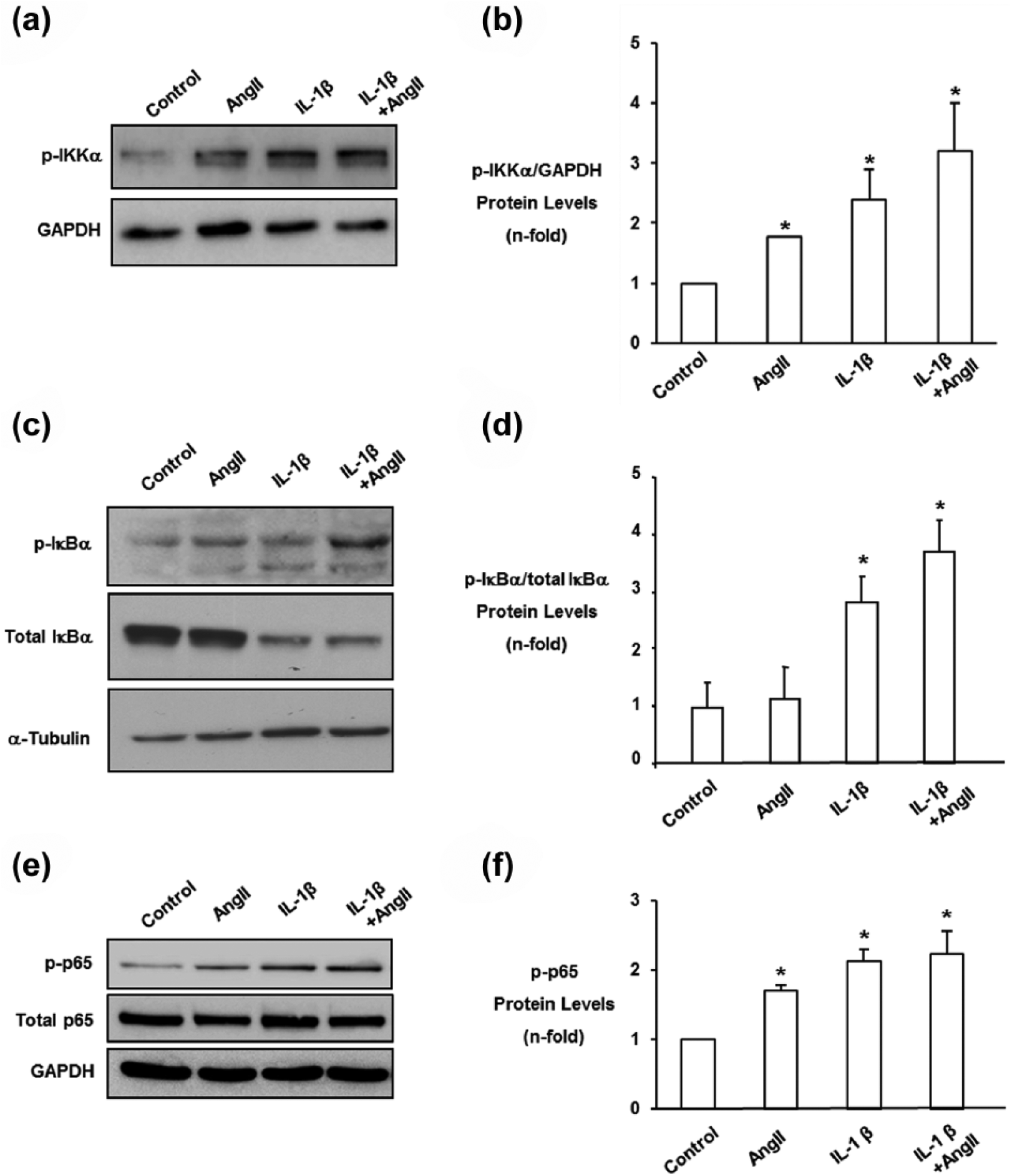
Angiotensin II (Ang II) and interleukin-1β (IL-1β) coincubation increased IKKα, IκBα and p65 nuclear factor-κB (NF-κB) phosphorylation, and diminished total IκBα protein levels in rat mesangial cells. Cells were coincubated for increasing times with 1 ng/ml IL-1β and 10−7 M Ang II, and total and cytosolic extracts were used. α-tubulin or GAPDH were used as loading controls. Part (a) shows a representative Western blot of phospho-IKKα and in (b) data corrected to GAPDH levels, as n-fold as mean±standard error of the mean (SEM) of four experiments. Figure (c) shows a representative Western blot of IκBα and phosphorylated-IκBα. Figure (d) shows the ratio of phosphorylated-IκBα versus total IκBα as mean±SEM of three experiments. Figure (e) shows a representative Western blot of phospho-p65 NF-κB and in (f) data corrected to GAPDH levels, as n-fold as mean±SEM of four experiments. *p<0.05 vs control.
NF-κB transcription factors consist of five distinct DNA-binding subunits, referred to as p50, p52, p65/Rel A, c-Rel, and Rel-B. We have analysed the NF-κB subunits in IL-1β and Ang II-treated cells, by a supershift assay using selective antibodies specifically for the NF-κB/Rel proteins, showing the involvement of p65/p50 heterodimer (data not shown). To further demonstrate the role of p65 NF-κB subunit in this process we have carried out confocal microscopy experiments. As shown in Figure 2(a), in unstimulated cells p65 NF-κB subunit is located in the cytosol, whereas after stimulation with IL-1β or Ang II p65 is translocated into the nuclei. The coincubation of both cytokines caused a synergistic effect on p65 nuclear localisation compared to each stimulus alone (Figure 2(a)). The active NF-κB complex is translocated into the nucleus where it binds to specific DNA sequences to regulate gene transcription. 18 IL-1β and Ang II caused a rapid activation of NF-κB DNA binding activity, observed by EMSA experiments after 30 min of stimulation, confirming previous data.16,21 When both stimuli were added together a synergistic effect on NF-κB DNA binding activity was found (Figure 2(b) and 2(c)). These data clearly demonstrate that the simultaneous presence of IL-1β and Ang II induced a synergistic activation of NF-κB pathway in cultured MCs.

Synergistic activation of nuclear factor-κB (NF-κB) caused by angiotensin II (Ang II) and interleukin-1β (IL-1β) in rat mesangial cells. Cells were coincubated with 1 ng/ml IL-1β and 10−7 M Ang II for 30 min. The p65 NF-κB subunit was localised by indirect immunofluorescence. Part (a) shows a representative confocal microscopy experiment of three done with similar results. (b) Nuclear extracts were obtained and NF-κB activity was determined by binding assay of nuclear proteins to an oligoconsensus labelled with γ- 32 [P]. The specificity of the reaction was established using competition assays with a 100-fold excess of unlabelled NF-κB oligonucleotide. The position of the NF-κB complexes and free-oligonucleotide is indicated. Part (b) shows a representative electrophoretic mobility shift assay (EMSA) experiment and in (c) data of DNA binding activity expressed as mean±standard error of the mean (SEM) of four experiments. *p<0.05 vs control; #p<0.05 vs Ang II; †p<0.05 vs IL-1β.
The simultaneous presence of IL-1β and Ang II causes overexpression of NF-κB-related inflammatory genes in mesangial cells
Coincubation of IL-1β and Ang II markedly increased MCP-1 gene expression (Figure 3(a)), and protein release (Figure 3(b)). Coincubation of IL-1β and Ang II caused a synergistic effect on IL-6 and VCAM-1 gene expression (Figure 3(a)). The involvement of NF-κB in the regulation of these genes was evaluated by using the NF-κB inhibitor, BAY 11-7082. Preincubation of MCs with BAY 11-7082 reduced proinflammatory gene overexpression observed by coincubation of Ang II and IL-1β (data not shown), and blocked MCP-1 release (Figure 3(b)).

(a) Cotreatment with interleukin-1β (IL-1β) and angiotensin II (Ang II) increased proinflammatory gene expression in mesangial cells. Cells were treated with 1 ng/ml IL-1β and 10−7 M Ang II for 18 h. Gene expression was evaluated by real-time PCR. (b) Cotreatment with IL-1β and Ang II increased monocyte chemoattractant protein-1 (MCP-1) release by a NF-κB mediated process. Some cells were pretreated with 10−5 M of BAY 11-7082, for one hour, before stimulation with 1 ng/ml IL-1β and 10−7 M Ang II for 24 h. Supernatants were collected and MCP-1 levels were evaluated by ELISA. Data are shown as mean±standard error of the mean (SEM) of four real-time PCR experiments and of three ELISA experiments. *p<0.05 vs control; #p<0.05 vs Ang II; †p<0.05 vs IL-1β; ¶p<0.05 vs IL-1β+Ang II.
AT1 receptor mediates the synergistic NF-κB activation and proinflammatory gene upregulation
To evaluate the Ang II receptor subtype involved in NF-κB synergistic activation, cells were preincubated with the AT1 receptor antagonist valsartan or AT2 receptor antagonist PD123319. Only treatment with valsartan, but not PD123319, markedly diminished the synergistic p65 translocation caused by coincubation with Ang II and IL-1β (Figure 4(a) and 4(b)). Finally, we evaluated the effect of AT1 blockade on proinflammatory gene expression. Only treatment with valsartan, but not PD123319, markedly diminished synergistic proinflammatory gene overexpression caused by coincubation with Ang II and IL-1β (Figure 4(c)). These data showed that synergistic effect on NF-κB pathway was mainly mediated through AT1 receptor.

Angiotensin receptor type 1 (AT1) blockade diminished (a) nuclear factor-κB (NF-κB) activation and (c) proinflammatory gene upregulation. Cells were pretreated for one hour with valsartan, AT1 receptor antagonist (10−5 M) or PD123319, AT2 receptor antagonist (10−5 M), then cells were stimulated with 1 ng/ml interleukin-1β (IL-1β) and/or 10−7 M Ang II for 30 min (a), (b) of 18 h (c). Part (a) shows a representative experiment of p65 NF-κB immunofluorescence staining and in (b) the immunofluorescence quantification as mean±standard error of the mean (SEM), of three different confocal microscopy experiments done. Part (c) shows percent of inhibition as mean±SEM of five real-time PCR experiments. *p<0.05 vs control; p<0.05 vs IL-1β+Ang II.
Ang II does not regulate IL-1β or its receptor in mesangial cells
In cultured MCs, Ang II did not regulate IL-1β mRNA levels or IL-1β receptor protein levels, at least, at the time points evaluated (Figure 5(a) and 5(b)).
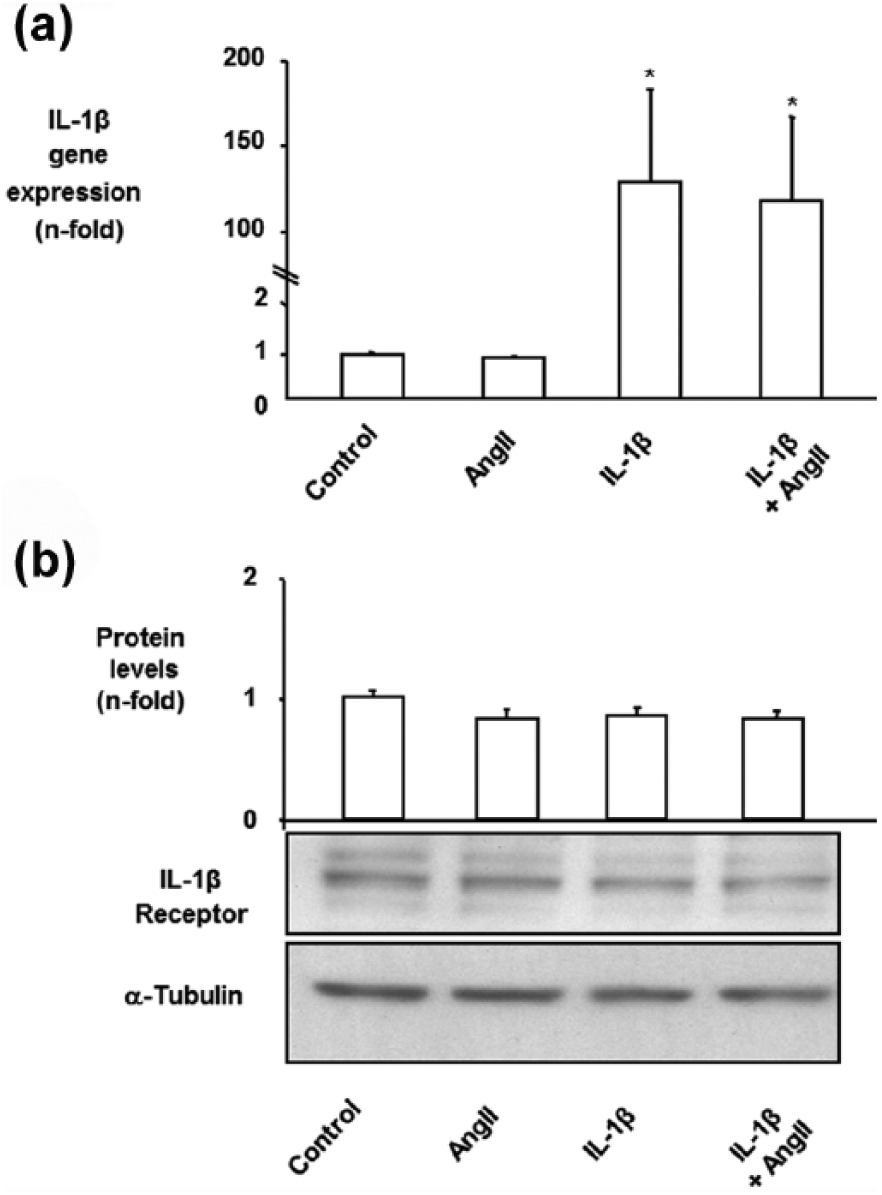
(a) Angiotensin II (Ang II) did not upregulate interleukin-1β (IL-1β) gene expression in cultured mesangial cells (MCs). MCs were treated with 1 ng/ml IL-1β and/or 10−7 M Ang II for 18 h. IL-1β gene expression was measured by real-time PCR. Figure shows data of mean±standard error of the mean (SEM) of three independent experiments. (b) IL-1β receptor protein levels in MCs. Cells were treated with 1 ng/ml IL-1β and/or 10−7 M Ang II for 24 h. Figure shows in top panel data of mean±standard error of the mean (SEM) of four experiments and in bottom a representative Western blot. α-Tubulin is used as loading control.*p<0.05 vs control.
Discussion
The major signalling pathway that controls inflammation-associated gene expression is the NF-κB pathway. Manyin vitro studies have demonstrated that proinflammatory cytokines such as Ang II and IL-1β activate the canonical NF-κB pathway in several cell types, including renal cells (mesangial, endothelial and tubuloepithelial cells).2,5,16,21 This canonical pathway is characterised by the activation of the IKK complex, 22 leading to the phosphorylation of IκBα and p65, and subsequent nuclear translocation of the active NF-κB complex, where it acts as a transcription factor. 19 In vivo studies have described how Ang II induced the phosphorylation of IKKα and IκBα, associated with NF-κB translocation to the nuclei and increased NF-κB activity in several tissues, including kidney,6,23 aorta 24 and heart. 25 In the kidney, Ang II increased p65 NF-κB phosphorylation at serine residue, 23 a key regulatory site in the COOH-terminal transactivation domain. 19 We have observed that in cultured MCs the simultaneous presence of IL-1β and Ang II caused a synergistic increase in the phosphorylation of IKK, IκBα and p65, as well as downstream effects, including p65 nuclear localisation and increased NF-κB DNA binding activity. There is a plethora of κB-dependent genes, predominantly regulating inflammation and cellular growth/survival. 26 Ang II induces many proinflammatory genes via NF-κB, and regulates renal inflammation through this pathway.2,16,21 We have found that coincubation of IL-1β and Ang II caused a marked overexpression of key NF-κB-controlled proinflammatory mediators, such as MCP-1 and IL-6. Moreover, blockade of NF-κB abrogates the synergistic effect of MCP-1 release caused by the coincubation of IL-1β and Ang II. Renal damage is associated with overexpression of a great variety of growth factors and cytokines, which influences the progression of kidney diseases. MCP-1 is an important molecule in renal injury that contributes to monocyte recruitment, promoting an inflammatory environment. 27 In human diabetic nephropathy activation of the canonical NF-κB pathway has been correlated to MCP-1 upregulation. 18 Moreover, increased urinary MCP-1 was associated with later stage disease of diabetic nephropathy. 28 Dysregulation of IL-6 activity or its signalling cascade has been implicated in the pathogenesis of a number of inflammatory diseases, 29 including chronic kidney disease. Our data support the hypothesis that increased MCP-1 and IL-6 by resident renal cell exposure to a combination of Ang II and IL-1β may lead to further attraction of inflammatory cells to the kidney, thereby generating an inflammatory loop that contributes to persistent renal inflammation.
In the kidney, depending on the cell type, a different receptor subtype, AT1 or AT2, is involved in Ang II-mediated NF-κB activation.6,30,31 In MCs, vascular smooth muscle cells, and in AT1/AT2 transfected COS-7 (monkey kidney fibroblast-like cell line) cells, Ang II activates the NF-κB pathway by both AT1 and AT2 receptors.5,24 In tubular epithelial cells NF-κB is activated by AT1 receptors while in glomerular endothelial cells it is AT2 mediated.5,6 The existence of the AT2/NF-κB pathway has been further demonstrated using specific AT2 agonists, AT1 knockout mice 24 or AT2 expressing cells. 5 The Ang II receptor subtype involved in inflammatory processes in the kidney is not completely elucidated. Experimental studies in models of renal injury have found that both AT1 and AT2 antagonists decreased NF-κB activation, as observed in the model of unilateral ureteral obstruction. 30 Moreover, in this model only combination of AT1 and AT2 antagonists blocked monocyte infiltration; NF-κB activation; and up-regulation of NF-κB–regulated proinflammatory genes, including MCP-1 and IL-6. 30 Data from AT2 knockout mice showed that the absence of AT2 signalling was associated with increased renal injury and mortality in chronic kidney disease. 32 In cultured cells Ang II via the AT1/NF-κB pathway upregulates MCP-1, IL-6 and VCAM-1.26,33,34 In glomerular endothelial cells Ang II via AT2 upregulates gene expression of RANTES (CCL5), another chemokine under NF-κB control. 35 In cultured MCs we have found that only the AT1, but not AT2, receptor antagonist abolished the synergistic effect on NF-κB activation and proinflammatory genes caused by coincubation of Ang II and IL-1β. Our data highlight the importance of the AT1/NF-κB pathway in the regulation of inflammatory response. Interestingly, in vascular cells IL-1β upregulated AT1 mRNA, increased AT1 binding after 24 h and enhanced Ang II–stimulated hypertrophic responses. 36 We observed that in MCs IL-1β also upregulated AT1 mRNA (data not shown), and enhanced proinflammatory-related events. Previous studies have reported the presence of NF-κB recognition sites in the 5’-flanking region of the rat AT1 receptor. 37 Moreover, the role of NF-κB in AT1 regulation has been demonstrated in cardiac fibroblasts exposed to TNFα. 38 Therefore another target of the AT1/NF-κB pathway could be the AT1 receptor.
In conclusion, there is substantial in vitro and in vivo evidence, demonstrated in part by our group, that binding of Ang II to AT1 receptors leads to the activation of the canonical NF-κB pathway as well as κB-dependent genes in intrinsic renal cells and the kidney.2,39 Many experimental studies have demonstrated that NF-κB inhibition ameliorates renal injury, including in Ang II-induced tissue damage,3,40 however the potential use of NF-κB inhibitors in human kidney disease is far from actual practice. Experimental studies have shown that blockade of the RAS diminished both renal NF-κB activation and inflammation.1,2 Data presented here show that AT1 blockade inhibits the amplification of the inflammatory response induced by the simultaneous presence of Ang II and the proinflammatory cytokine IL-1β, by the modulation of the NF-κB pathway. These data suggest that in human renal inflammatory conditions, beneficial effects of treatment with AT1 antagonists, besides its well-known effects on blood pressure regulation, could exert additional anti-inflammatory properties by modulating Ang II-induced pathological effects, including cytokine-induced NF-κB activation.
Footnotes
Acknowledgements
The authors wish to express their thanks for the technical help of Mar Gonzalez Garcia-Parreño in confocal microscopy.
Conflict of interest
None declared.
Funding
This work was supported by grants from the Instituto de Salud Carlos III (ISCIIIRETIC REDinREN RD06/0016, RD12/0021, PI11/01854), Comunidad de Madrid (Fibroteam S2010/BMD-2321, S2010/BMD-2378), and Research Institute Queen Sophia (IRSIN). Programa Intensificación Actividad Investigadora (ISCIII/Agencia Laín-Entralgo/CM) to AO. MA and ESL are supported by a ‘Sara Borrell’ postdoctoral contract from Instituto de Salud Carlos III (CD10/00347 and CD09/00066, respectively).