
Abstract
Introduction:
In this study, we investigated whether AngII receptors (AT1a and AT2) contributed to the development of the aldosterone-induced inflammatory response of rat mesangial cells (RMCs).
Materials and methods:
RMCs were isolated from the glomeruli of normal or diabetic rats which were produced by injection of streptozotocin, and cultured in high-glucose media. In order to evaluate the effects of aldosterone, the expression of AT1a, AT2, NF-κB and MCP-1 was detected. In addition, in order to evaluate the role of Ang II receptors, AT1a and AT2 genes were blocked and the expression of NF-κB and MCP-1 was detected. Moreover, for assessing the relationship between NF-κB and MCP-1, the NF-κB gene was blocked and MCP-1 expression was detected.
Results:
Aldosterone significantly increased AT1a, AT2, NF-κB and MCP-1 levels in RMCs in a dose-dependent manner, whereas eplerenone (EPI), a selective aldosterone antagonist, partly inhibited the effects of aldosterone. When AT1a and AT2 genes were blocked, the expression of NF-κB and MCP-1 was greatly inhibited. Moreover, when the NF-κB gene was silenced, the expression of MCP-1 was reduced.
Conclusion:
We demonstrated that aldosterone induced an inflammatory response in RMCs cultured in high-glucose media via the AT1a and AT2 pathways.
Introduction
Mesangial cells (MCs) are the most active glomerular intrinsic cells, and synthesize and secrete a large number of extracellular matrices and numerous cytokines in glomerular disease.1,2 They are thought to be one of the main target cells of various pathogenic factors, and can proliferate, synthesize and secrete a variety of inflammatory mediators, resulting in a kidney inflammatory response, promoting the progress of diabetic nephropathy (DN). Therefore, studying the dysfunction of MCs is of great significance in exploring the pathogenesis of DN.
Aldosterone plays a pivotal role in the pathogenesis of renal disease, including DN. 3 Animal studies have confirmed that mineralocorticoid receptors are present in the renal tubule and partial glomerular cells. 4 One study showed that along with angiotensin-converting enzyme inhibitors, aldosterone also causes cardiovascular and renal damage, and spironolactone (even with injection of Ang II) can exert obvious cardiovascular and renal protective roles. Moreover, many research data have shown that the aldosterone system, inhibited in DN, has a beneficial effect independent of blockade of the renin–angiotensin system.5–7
Ang II mainly acts through binding to two specific receptors: Ang II type 1a (AT1a) and Ang II type 2 (AT2) receptors. AT1 is responsible for most of the pathophysiological effects of Ang II by promoting proliferation, inflammation, and fibrosis. 8 However, the role of AT2 has not been completely defined. Previous studies have shown that AT2 is involved in inhibition of cell growth and inflammatory cell recruitment in the kidney.9,10 However, there are emerging data that AT2 also produces proinflammatory effects and is involved in promotion of fibrosis and hypertrophy.11,12 The antagonist of AT2, but not AT1, can diminish the number of inflammatory cells in different animal models such as systemic infusion of Ang II and unilateral ureteral obstruction. 13 Growing evidence has shown that aldosterone can influence the signaling or trafficking of the Ang II receptor. Indeed, mineralocorticoids such as deoxycorticosterone and aldosterone cause up-regulation of Ang II binding to blood vessels and cultured vascular smooth muscle cells. Several studies have unraveled further evidence of a crosstalk between AT1 and mineralocorticoid receptor (MR). These studies suggest that vascular responses to Ang II can be mediated via direct signaling crosstalk between MR and AT1. 14
Based on the extensive clinical and animal model studies cited above, we hypothesize that AT1a/AT2 interact with MR in intracellular signaling. We seek to understand more precisely the molecular mechanisms underlying crosstalk between AT1a/AT2 and MR in the aldosterone-induced inflammatory reaction of DN.
Materials and methods
Experimental animals and cell culture
Diabetes was induced in male Wistar rats (175–200 g, 8 weeks) by 65 mg/kg streptozotocin (STZ, dissolved in 0.1 M citrate buffer, pH 4.5) i.v. Animals were considered diabetic if blood glucose concentration increased to >15 mmol/l within 72 h after STZ injection and remained elevated. Rat mesangial cells (RMCs) were isolated from the glomeruli of rats (diabetic and healthy) using a differential sieving technique. Briefly, cortices were removed and minced before passage through a series of steel sieves with decreasing pore sizes (200, 150 and 75 µm). Isolated glomeruli were then digested with collagenase. Digested glomeruli were cultured in RPMI 1640 combined with antibiotics (100 U/ml penicillin and 10 µg/ml streptomycin) and 20% fetal bovine serum (FBS; Sigma Chemical Co., St Louis, MO) until MCs reached confluence. The cells were passaged using trypsin/EDTA, and cells between passages 3 and 8 were used for experiments. Cells were characterized, and the homogeneity of the MCs was assessed by identifying typical morphology and patterns of staining with antibodies against smooth muscle cell-specific actin, factor VIII antigen, leukocyte common antigen and by the presence of hillocks. 15 RMCs were cultured in RPMI 1640 medium with 10% FBS at 37°C in a 5% CO2 incubator. Then the cells were seeded in a 100 mm culture dish for western blot measurement and a 6-well culture plate for qPCR. Near-confluent RMCs were incubated with serum-free media for 24 h to arrest and synchronize the cell growth. After this time period, the supernatant was removed and the media was changed to fresh serum-free RPMI 1640 containing high-concentration glucose (30 mmol/l), dexamethasone (10-6mol/l) and different concentrations of aldosterone (ALD, 10-5–10-9mol/L). In some experiments, cells were pretreated with eplerenone (EPI, 10-5mol/l), losartan (10-4mol/l), PD123319 (10-5mol/l) or RU486 (10-5mol/l) for 3 h before the addition of aldosterone. The high-glucose media group contained 30 mmol/l glucose only. However, the normal glucose media group contained 5.6 mmol/l glucose and 24.4 mmol/l mannitol.
All the animal research protocols used in this study were approved by the Animal Ethics Review Committees of Harbin University and accorded with the principles stated in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication).
Gene knockdown
Silencing of AT1a, AT2 and NF-κB genes with siRNA (Santa Cruz Biotechnology, Inc.) was performed as per the instructions of the manufacturer. Cells were cultured in antibiotic-free growth medium to 60% confluence. A transfection mixture of AT1a, AT2 or NF-κB siRNA (60 nM) and transfection reagent (0.5ul/ml) was made in RPMI-1640 reduced serum medium. Cells were incubated with the transfection mixture in complete medium for 48 h. This protocol was optimal for silencing AT1a, AT2 and NF-κB and resulted in 95% cell survival. After transfection, cells were incubated in growth medium for 24 h and then stimulated with aldosterone for 12 and 24 h. Controls were treated with scrambled siRNA and transfection reagent.
Quantitative polymerase chain reaction (qPCR) analysis
RMCs were treated for 12 h in accordance with the above methods, then total RNA was extracted from experimental cells with Trizol reagent according to standard procedures (Invitrogen, American). qPCR was performed using the Roche LightCycler 480II System (Roche Diagnostics). The Reverse Transcription Kit (Invitrogen, American) was used to generate cDNA, and SYBR Assays (TaKaRa, Japan) was used to quantitatively detect mRNAs. The following primer oligonucleotide sequences were used: AT1a (forward: 5’-CTGGCAGAAATGCAATCTCA-3’; reverse: 5’-ACAAAGGTTCCTTGCCCTTT-3’; 158 bp product expected); AT2 (forward: 5’-CCCTGGCAAGCATCTTATGT-3’; reverse: 5’-CCAGCAGACCACTGAGCATA-3’; 160 bp product expected); NF-κB (forward: 5’-AACACTGCCGAGCTCAAGAT-3’; reverse: 5’- CATCGGCTTGAGAAAAGGAG-3-3’; 162 bp product expected); MCP-1 (forward: 5’-ATGCAGTTAATGCCCCACTC-3’; reverse: 5’-TTCCTTATTGGGGTCAGCAC-3’; 166 bp product expected); and β-actin (forward: 5’-CGCCAACCGCGAGAAGAT-3’; reverse: 5’-CGTCACCGGAGTCCATCA-3’; 168 bp product expected). β-actin was used as endogenous reference gene for normalization. The relative expression levels were determined using the comparative threshold cycle (2-△△Ct) method as described by the manufacturer.
Protein extraction and western blotting analyses
The protein expression of AT1a, AT2, NF-кB and MCP-1 was measured by western blotting. After incubation with aldosterone for 24 h, whole-cell lysates extracted from the cultured RMCs were lysed in SDS sample buffer, and total protein was extracted from this homogenate. The protein concentration in each sample extract was measured using a protein assay kit, and was then adjusted to the same value in all samples with 2× 4% SDS sample buffer. The samples were boiled for 5 min followed by loading on a 10% SDS-PAGE gel (30 μg protein/10 ml per well) for electrophoresis using a Bio-Rad mini gel apparatus at 35 mA/gel for 45 min. The fractionated protein on the gel was transferred onto a PVDF membrane (Millipore) and electrophoresed at 300 mA for 90 min. The blots were incubated in 5% nonfat dry milk for 1 h at room temperature and then incubated with the indicated antibodies (AT1a rabbit polyclonal IgG, AT2 rabbit polyclonal IgG, NF-кB p65 rabbit polyclonal IgG and MCP-1 rabbit polyclonal IgG, Abcam HK Ltd.) for 2 h. After three washes in 0.1% Tween 20 in PBST, the blots were incubated with secondary antibodies for 1 h and then treated with enhanced chemiluminescence substrate for 5 min at room temperature. The bands in the membrane were visualized and analyzed using UVP BioImaging Systems. The final reported data were the normalized AT1a, AT2, NF-кB and MCP-1 band densities by β-actin.
Statistical analyses
All results are presented as mean ±SEM. Analysis of variance (ANOVA) was used to assess the differences between multiple groups by a microcomputer-assisted program with SPSS for Windows 17.0; p < 0.05 was considered to be statistically significant.
Results
The differential expression of MR, AT1a, AT2, MCP-1 and NF-KB in RMCs from normal and diabetic rats
To test which MC might represent a suitable cell to study the role of AT1a and AT2 in proinflammatory cytokine production, we determined the mRNA expression of MR, AT1a, AT2, MCP-1 and NF-KB using qPCR. RMCs from normal or diabetic rats were cultured in 5.6 or 30 mmol/l glucose medium for 12 h and 24 h. Substantial levels of mRNA expression for all five genes were detected. The mRNA expression of RMCs cultured in 30 mmol/l glucose medium increased obviously, compared with normal glucose media group (5.6 mmol/l)(Figure 1(a)–1(e)). However, in the high-glucose media group (30 mmol/l), the mRNA expression of diabetic RMCs increased more obviously, compared with normal RMCs (Figure 1(a)–1(e)). The expression pattern suggested that RMCs from the diabetic model rats were fit for investigating mechanisms of inflammatory reaction. Thus, we chose these cells to complete our study.
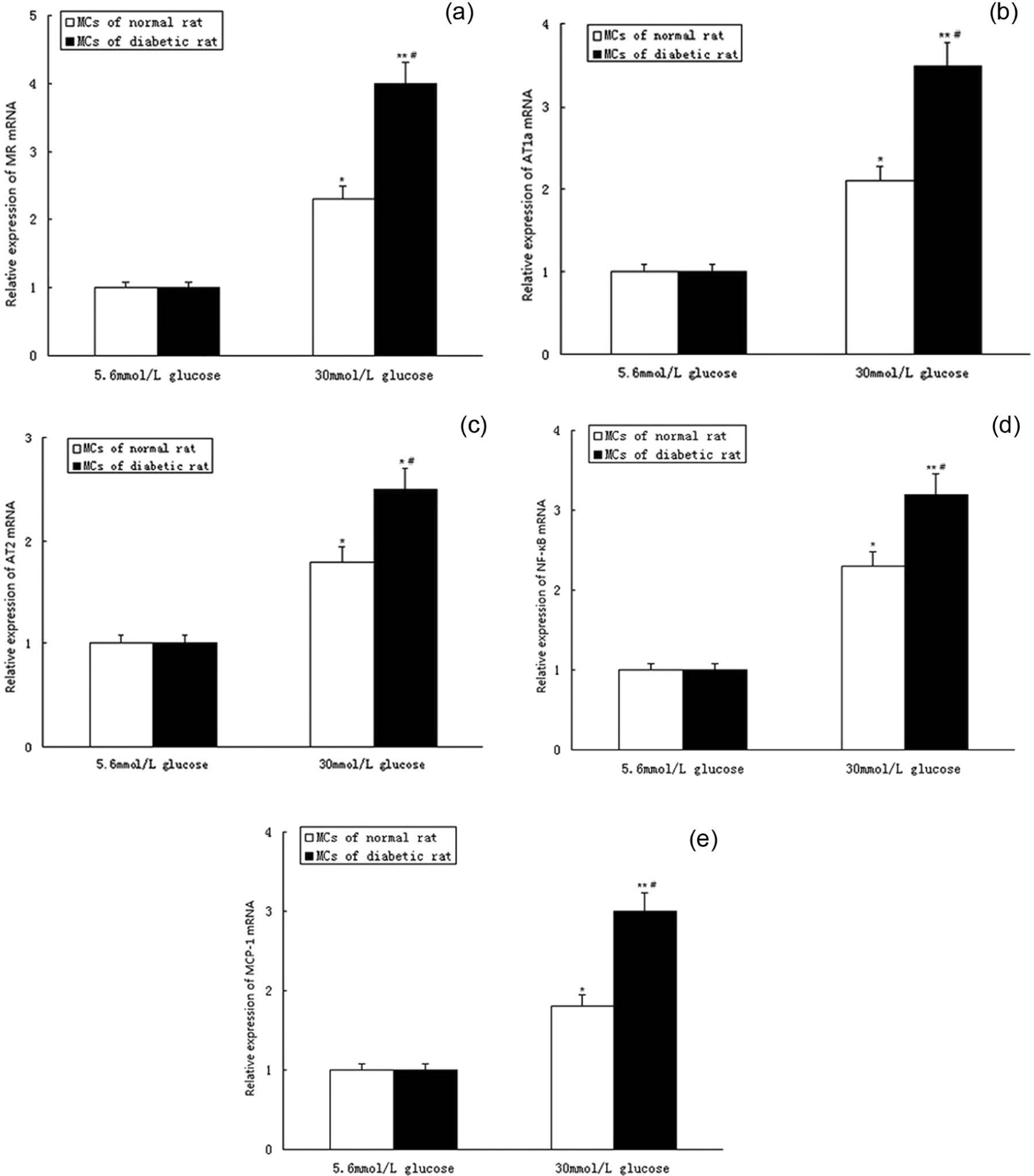
The different expression of MR, AT1a, AT2, MCP-1 and NF-KB in RMCs from normal and diabetic rats. Panel A–G shows MR, AT1a, AT2, NF-κB and MCP-1 mRNA expression: RMCs were cultured with different concentration of glucose (5.6 and 30 mmol/l) for 12 h. *p<0.5, **p<0.01,*** p<0.001, versus the normal glucose media group (5.6 mmol/l); #p<0.5, versus normal RMCs cultured in the high-glucose media group (30 mmol/l). RMCs: rat mesangial cells; MR: mineralocorticoid receptor; AT1a: angiotensin II type 1a receptor; AT2: angiotensin II type 2 receptor; MCP-1: monocyte chemoattractant protein-1.
Effects of aldosterone on AT1aR and AT2R expression
RMCs from diabetic rats were exposed to aldosterone (10-5–10-9mol/l) for 12 and 24 h, respectively. The effects of aldosterone on AT1a and AT2 mRNA and protein expressions are shown in Figure 2. Aldosterone significantly induced the expression of AT1a and AT2 mRNA (Figure 2(a), Figure 2(b)) and protein (Figure 2(e), Figure 2(f)) of RMCs cultured in high-glucose media in a dose-dependent manner. Since aldosterone can also act through non-genomic pathways, we also performed shorter time stimulations (5, 10, 30, 60, 120 minutes).We found that the expression of Ang II receptors gradually increased. When RMCs were exposed to aldosterone (10-7mol/l) for 30 min, the expression of AT1a and AT2 mRNA increased obviously (Figure 2(g)).

Effect of aldosterone on AT1a, AT2, NF-κB and MCP-1 expression of RMCs cultured in high-glucose media. Panel A–D shows AT1a, AT2, NF-κB and MCP-1 mRNA expression: cells were cultured with aldosterone (10-5–10-9mol/l) for 12 h. Panel E–F shows AT1a, AT2, NF-κB and MCP-1 protein expression: cells were cultured with aldosterone (10-5–10-9mol/l) for 24 h. *p<0.5, **p<0.01,*** p<0.001. RMCs: rat mesangial cells; ALD: aldosterone; AT1a: angiotensin II type 1a receptor; AT2: angiotensin II type 2 receptor; MCP-1: monocyte chemoattractant protein-1.
Effect of aldosterone on NF-κB expression
In this experiment, diabetic RMCs were exposed to aldosterone (10-5–10-9mol/l) for 12 and 24 h. The effect of aldosterone on NF-кB expression was detected by qPCR and western blotting. We found that aldosterone could increase the expression of NF-кB in a dose-dependent manner (Figure 2(c), Figure 2(e), Figure 2(f)). Eplerenone, a selective aldosterone antagonist, could partially block the effect of aldosterone (Figure 3(a)–3(d)), compared with the aldosterone (10-7mol/l) group, p<0.05.
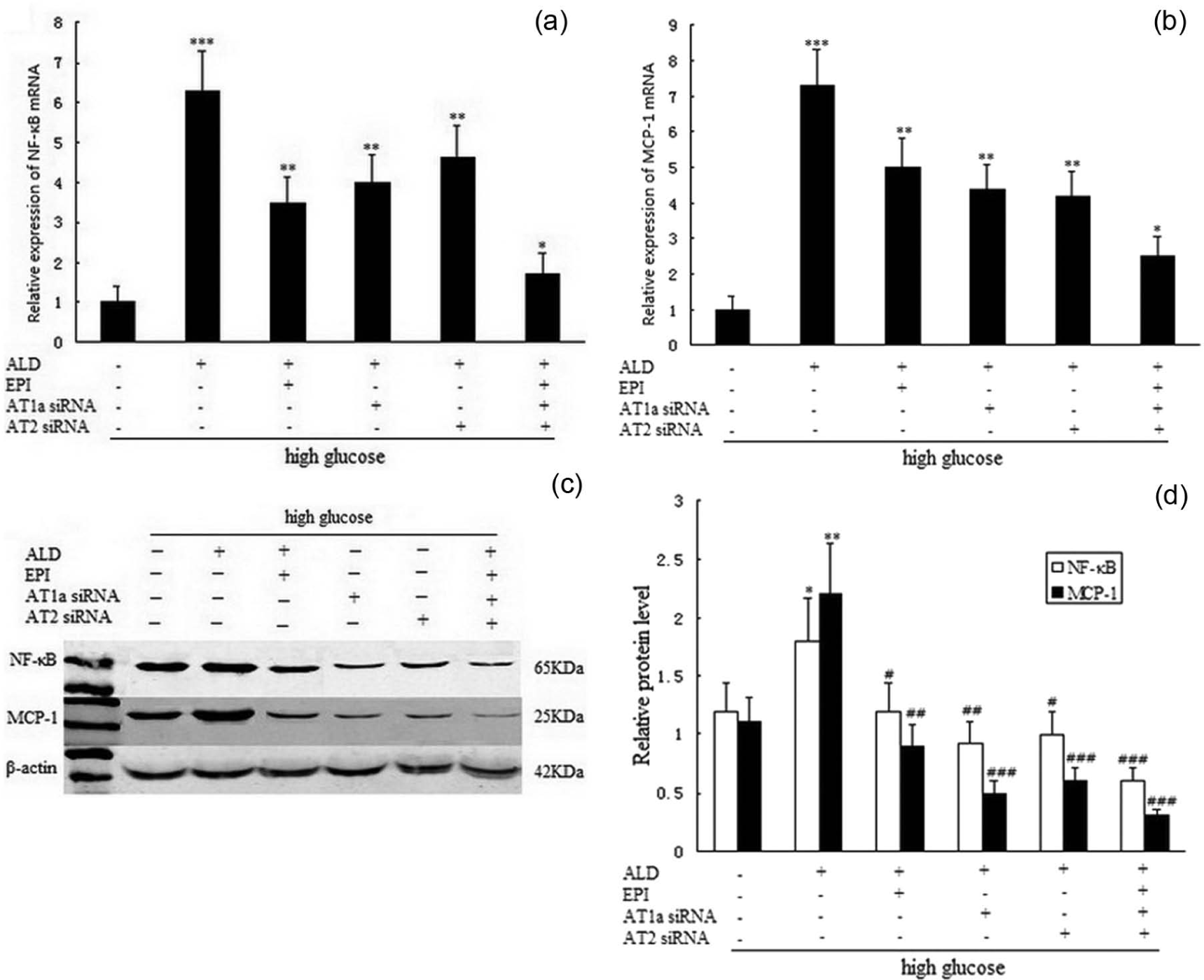
Effect of AT1a siRNA and AT2 siRNA on NF-κB and MCP-1 expression of RMCs cultured in high-glucose media. Panel A–B shows NF-κB and MCP-1 mRNA expression. Panel C–D shows NF-κB and MCP-1 protein expression. * p<0.5,** p<0.01,*** p<0.001, versus high-glucose media group (30mmol/l); # p<0.5, ## p<0.01, ### p<0.001, versus aldosterone (10-7mol/l) group. RMCs: rat mesangial cells; ALD: aldosterone; EPI: eplerenone; AT1a: angiotensin II type 1a receptor; AT2: angiotensin II type 2 receptor; MCP-1: monocyte chemoattractant protein-1.
Effect of aldosterone on MCP-1 expression
When the RMCs from diabetic model rats cultured in high-glucose media were exposed to aldosterone (10-5–10-9mol/l) for 12 and 24 h, we observed overproduction of MCP-1 mRNA and protein in a dose-dependent manner (Figure 2(d)–2(f)). Eplerenone significantly inhibited the expression of MCP-1 compared with the aldosterone group (10-7mol/l), p<0.01 (Figure 3(a)–3(d)).
Effect of gene knockdown of AT1a and AT2 on NF-κB and MCP-1 expression
The diabetic RMCs cultured in high-glucose media, after silencing of the AT1a and AT2 genes with siRNA, were exposed to aldosterone (10-7mol/l) for 12 and 24 h. We observed that the expression of NF-кB and MCP-1 mRNA and protein was down-regulated, compared with the high-glucose group (Figure 3(a)–3(d)). However, the expression of NF-кB and MCP-1 was significantly inhibited in the combination of EPI, AT1a siRNA and AT2 siRNA group, compared with high-glucose group (Figure 3(a)–3(d)) p<0.001.
Effect of antagonists of AT1a and AT2 on NF-κB and MCP-1 expression
Diabetic RMCs cultured in high-glucose media, which were incubated with EPI (10-5mol/l), losartan (10-4mol/l) and PD123319 (10-5mol/l) for 3 h, were exposed to aldosterone (10-7mol/l) for 12 and 24 h. We observed that the expression of NF-кB and MCP-1 mRNA and protein was down-regulated, compared with the normal glucose (5.6 mmol/l) or high-glucose (30 mmol/l) group (Figure 4(a)–4(c)). However, the combination Ang II; receptor antagonists and aldosterone receptor blocker significantly inhibited the expression of NF-кB and MCP-1. The diabetic RMCs cultured in high-glucose media were exposed to EPI (10-5mol/l), losartan (10-4mol/l) and PD123319 (10-5mol/l) individually for 12 and 24 h. The expression of NF-кB and MCP-1 mRNA and protein was reduced compared with the high-glucose group (30mmol/l) (Figure 4(a), Figure 4(d) and Figure 4(e)).
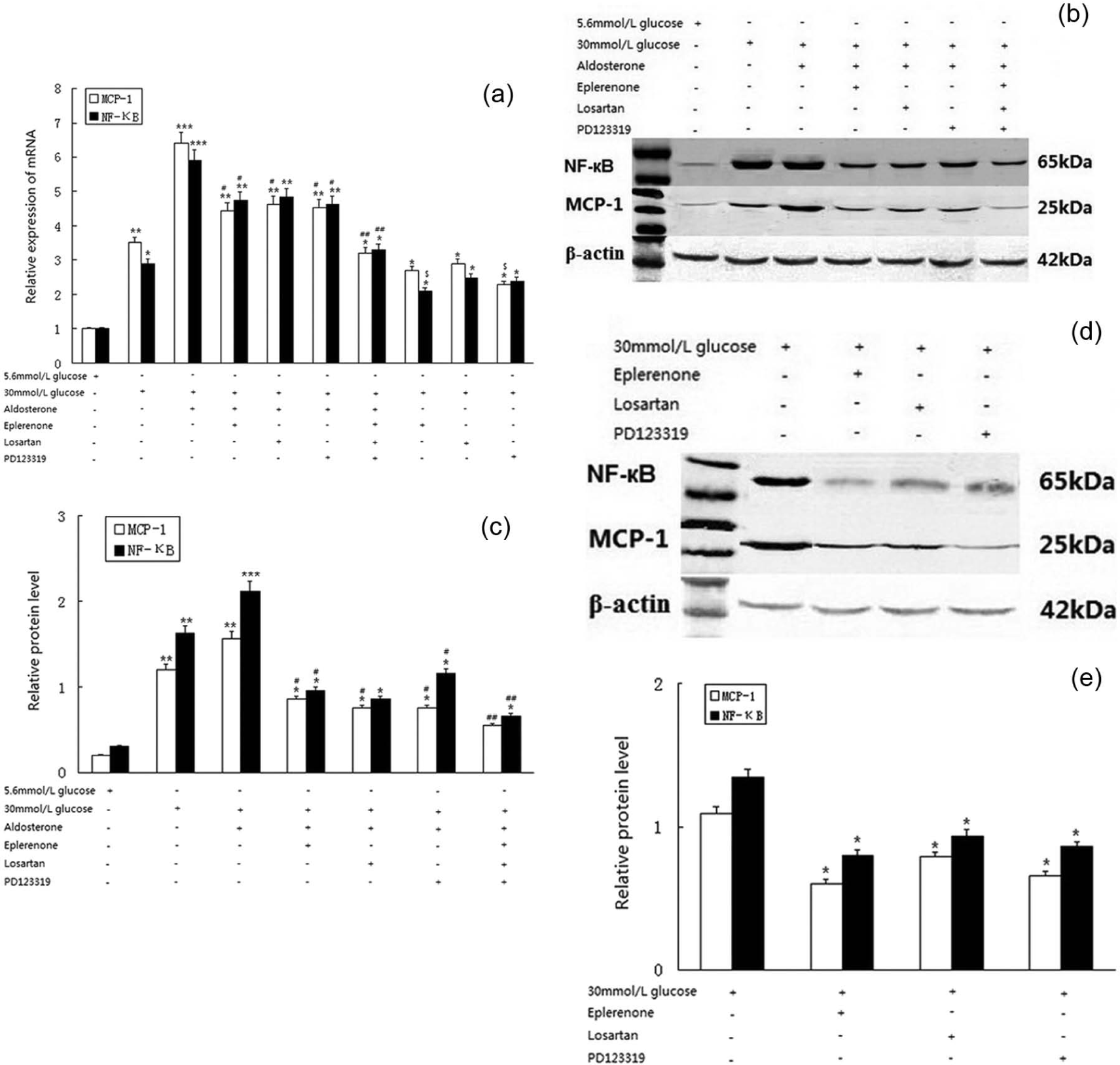
Antagonists of AT1a and AT2 on NF-κB and MCP-1 expression. Panel A shows NF-κB and MCP-1 mRNA expression. Panel B–E shows NF-κB and MCP-1 protein expression. * p<0.5,** p<0.01,*** p<0.001, versus the normal glucose media group (5.6mmol/l); # p<0.5, ## p<0.01, versus the aldosterone (10-7mol/l) group. $ p<0.5 versus the high-glucose media group (30 mmol/l), AT1a: angiotensin II type 1a receptor; AT2: angiotensin II type 2 receptor; MCP-1: monocyte chemoattractant protein-1; losartan: angiotensin II type 1 receptor blocker; PD123319: angiotensin II type 2 receptor blocker.
Effect of gene knockdown of NF-κB on MCP-1 expression
RMCs cultured in high-glucose media were stimulated by aldosterone (10-7mol/l) for 12 and 24 h. The expression of MCP-1 mRNA and protein was significantly up-regulated compared with the high-glucose group (p<0.001). When the NF-кB gene was silenced with siRNA, RMCs were exposed to aldosterone (10-7mol/l) for 12 and 24 h. We observed that the overproduction of MCP-1 was down-regulated compared with the aldosterone group (Figure 5(a)–5(c)).
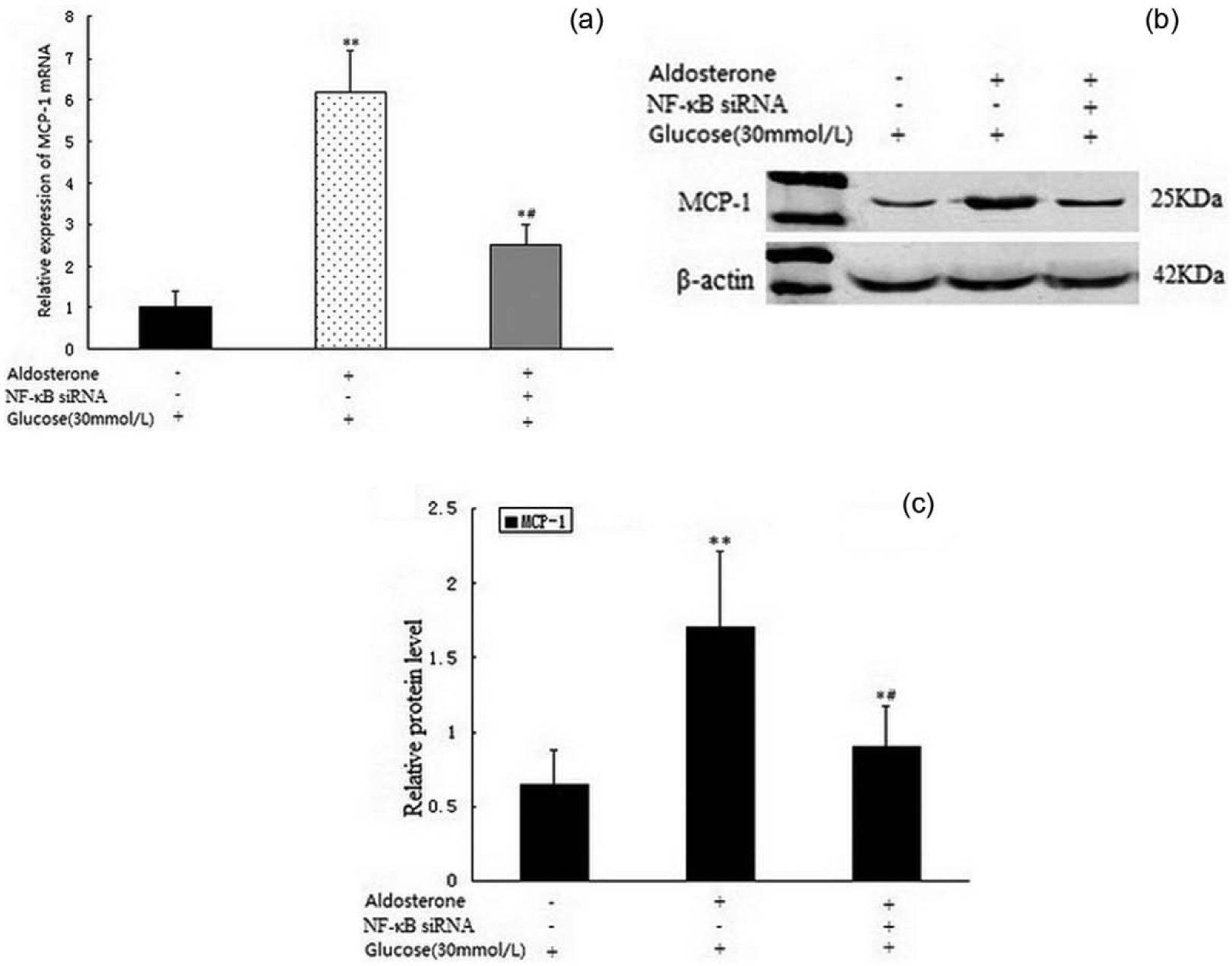
Effect of NF-κB siRNA on MCP-1 expression of RMCs cultured in high-glucose media. Panel A shows MCP-1 mRNA expression. Panel B–C shows MCP-1 protein expression. * p<0.5, ** p<0.001, versus the high-glucose media group (30mmol/l); # p<0.01 versus the aldosterone (10-7mol/l) group. RMCs: rat mesangial cells; MCP-1: monocyte chemoattractant protein-1.
The effect of dexamethasone on the expression of inflammatory factors
Dexamethasone inhibited the inflammatory response. In this experiment, RMCs cultured in high-glucose media were exposed to dexamethasone (10-6mol/l) for 12h and 24h, and the expression of NF-кB and MCP-1 was decreased compared with the normal glucose group (5.6 mmol/l) or aldosterone group (10-7mol/l). The effect of dexamethasone was abolished by the glucocorticoid receptor antagonist RU486. When RMCs were stimulated by aldosterone and RU486, the expression of NF-кB and MCP-1 increased (Figure 6(a)–6(c)).
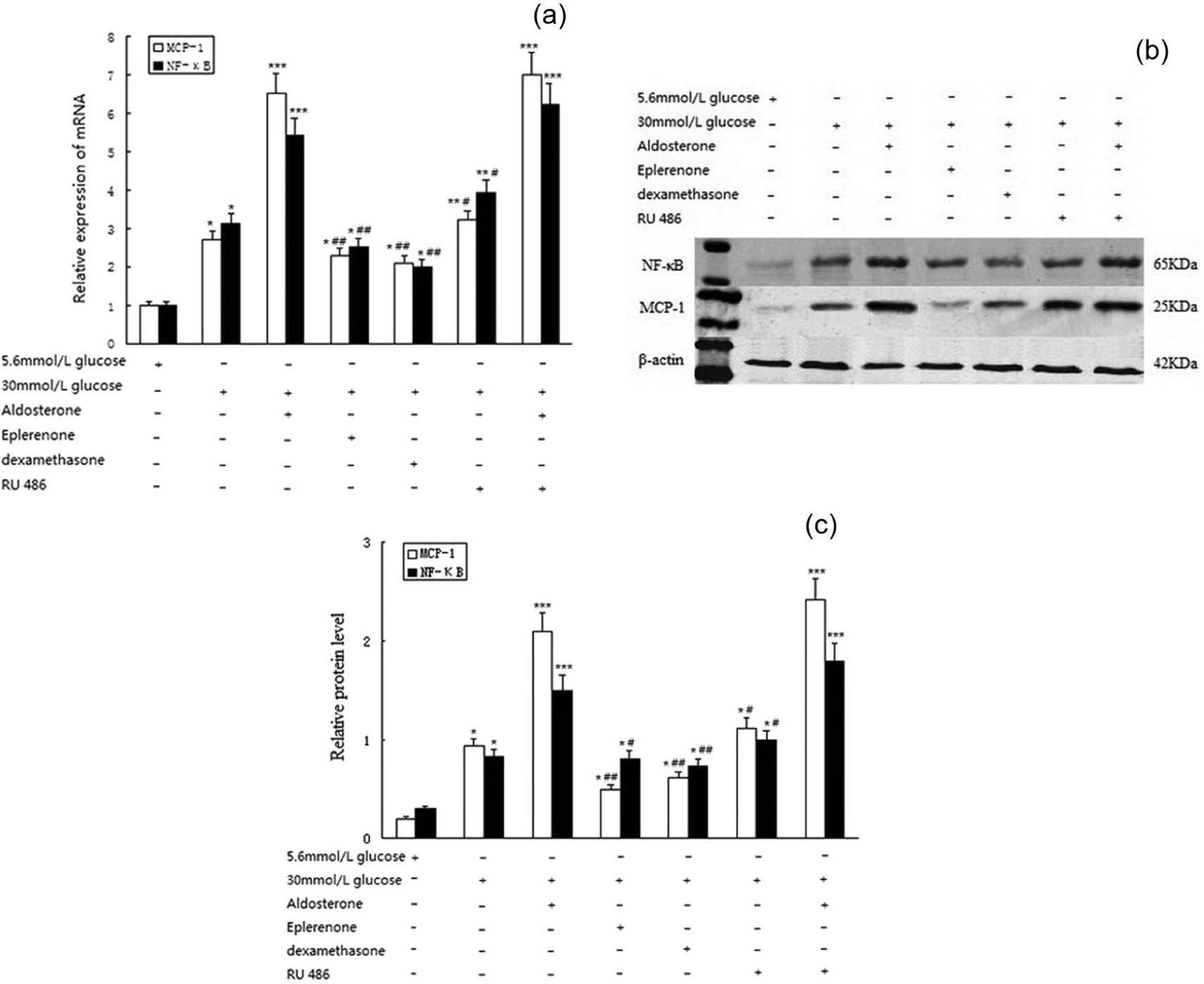
The effect of dexamethasone on inflammatory factor expression. Panel A shows NF-κB and MCP-1 mRNA expression. Panel B–C shows NF-κB and MCP-1 protein expression.*p<0.5, **p<0.01, ***p<0.001, versus normal glucose media group (5.6mmol/l); # p<0.5, ## p<0.01, versus the aldosterone (10-7mol/l) group. MCP-1: monocyte chemoattractant protein-1; RU486: glucocorticoid receptor antagonist.
Discussion
This study has shown that aldosterone can up-regulate NF-κB and MCP-1 expression via AT1a/AT2 pathway of RMCs cultured in high-glucose media. AT2 also participated in the inflammatory reaction. The inflammatory response plays an important role in the pathogenesis of DN.16,17 Some researchers believe that aldosterone can induce the inflammatory response via the NF-κB/MCP-1 pathway and participate in the development of DN. 18 They have speculated that aldosterone may be an important proinflammatory factor which can induce the initial inflammatory response.19–22
In this study, we found that aldosterone could up-regulate the AT1a and AT2 expression of RMCs cultured in high-glucose media in a dose-dependent manner. At present, it is widely recognized that the activation of AT1a in MCs can stimulate the secretion of multiple inflammatory factors and growth factor, resulting in kidney damage. In the present study, EPI down-regulated the expression of inflammatory factors in RMCs.
At present, the specific function of AT2 is not fully understood. Previous studies have shown that the activation of AT2 could inhibit the roles of AT1a, consequently inhibiting cells differentiation, proliferation and growth. However, some research has shown that AT2 participates in the progress of kidney disease. Moulder et al. 23 have shown that treatment with AT1a or AT2 antagonist can delay the progress of experimental radiation nephropathy. They proved that the relationship between AT1a and AT2 was not simply contradictory.
It has been reported that the adoption of an AT2 blocker could reduce the infiltration of inflammatory factors into glomerular and tubulointerstitial areas, playing an anti-inflammatory role. Therefore, we speculated that AT2 was involved in the inflammatory response of kidney injury. Previously, Esteban et al. 24 had reported that AT1a and AT2 might be involved in a common signaling pathway, such as stimulating the activation of NF-κB and inducing inflammation. Furthermore, the expression of AT2 was increased in a compensatory manner when AT1a was blocked. Thus AT2 might replace the role of AT1a in participating in the progress of renal disease. 24 Accordingly, we speculated that aldosterone was involved in the development of DN via AT2-induced inflammation, with crosstalk between AT1a and AT2.
MCP-1 participates in the development of glomerular inflammation through activation of monocytes and macrophages, which can be produced by glomerular MCs. 17 Aldosterone induces MCP-1 expression in heart tissue, which is mediated by NF-кB.25,26 Our experimental results showed that the administration of aldosterone could induce NF-κB and MCP-1 overproduction via AT1a/AT2. However, treatment with eplerenone or gene knockdown could reduce the effects of aldosterone. Therefore, we speculated that aldosterone might regulate the inflammatory response through the AT1a/AT2 pathway.
In contrast to the aldosterone-mediated inflammatory action, dexamethasone strongly inhibited the expression of the inflammatory factors. Our preliminary research data provided evidence that dexamethasone had an anti-inflammatory action in RMCs cultured in high-glucose media. However, there might be interaction between aldosterone and glucocorticoids. A disorder of the balance between the glucocorticoid receptor and the mineralocorticoid receptor could contribute to the inflammatory actions of MCs. The exact mechanisms involved are still unknown, and need further research.
In conclusion, we have demonstrated that aldosterone contributed to up-regulate the expression of AT1a and AT2, and induce the inflammatory reaction partly via AT1a/AT2-NF-κB/MCP-1 pathway in glomerular MCs cultured in high-glucose media. AT2 may be involved in the inflammatory reaction. The exact mechanism should be further defined.
Footnotes
Conflict of interest
None declared
Funding
This work was supported by the International Cooperation of Science and Technology Office [grant number WC05C05].