
Abstract
Introduction:
The insulin-regulated aminopeptidase (IRAP) is expressed in several cell types, where it is mainly located in specialized secretory endosomes that are quickly recruited to the cell surface upon cell type-specific activation. Here we describe for the first time the expression and subcellular distribution of IRAP in macrophages.
Methods:
IRAP mRNA expression, protein expression and presence at the cell surface was investigated by real-time polymerase chain reaction (PCR), Western blot and [3H]IVDE77 binding, respectively.
Results:
IRAP mRNA expression was increased by interferon-γ (IFN-γ) and lipopolysaccharide (LPS), but not by anti-inflammatory cytokines (interleukin (IL)-4, IL-10, transforming growth factor β (TGF-β)). IFN-γ increased [3H]IVDE77 binding steadily over time, while LPS quickly and transiently recruited IRAP to the cell surface. Combined stimulations with IFN-γ and LPS showed the same pattern as LPS alone. Latex particles also induced a transient recruitment of IRAP to the cell surface, but no difference was observed in phagocytic uptake between wild-type and IRAP–/– macrophages, suggesting that the enzymatic activity of IRAP is not required for the ingestion of particles.
Conclusion:
IRAP is more highly expressed in pro-inflammatory M1-activated macrophages and its presence at the cell surface is modulated upon exposure to IFN-γ, LPS or exogenous particles.
Introduction
The insulin-regulated aminopeptidase (IRAP, EC 3.4.11.3) is a membrane-bound zinc-dependent aminopeptidase found in diverse tissues, including heart, brain and fat. 1 IRAP has also been described in various immune cells, such as dendritic cells (DCs), mast cells, B cells and T cells.2,3 However, to the best of our knowledge there is little information either on its presence in macrophages or on its regulation in leukocytes. Macrophages are tissue-resident white blood cells that regulate and actively participate in inflammation. 4 These cells typically express high levels of proteases and peptidases,5,6 in line with their function in phagocytosis and extracellular matrix degradation. 7 One example is aminopeptidase-N (AP-N, CD13, EC 3.4.11.2), which participates in multiple functions in macrophages including antigen trimming, phagocytosis, cell adhesion and signal transduction.8,9 IRAP and AP-N are structurally closely related and share to some extent common substrate specificity.10,11 IRAP has a broad pH and substrate range, with a preference for (but not limited to) Cys-rich peptides such as vasopressin, oxytocin and somatostatin. 12 It also helps in the generation of antigenic peptides from exogenous antigens for cross-presentation on major histocompatibility complex (MHC) class I in DCs. 3 But what sets IRAP apart from the other members of the M1 aminopeptidase family, is its extended cytoplasmic domain (109 aa). 13 This N-terminal domain contains two di-leucine trafficking motifs (preceded by acidic residues), which are responsible for the correct localization of IRAP and its complex intracellular trafficking.14,15 In this respect, IRAP is mainly located perinuclearly in specialized secretory endosomes under basal conditions.2,16,17 These endosomes can be quickly recruited to the cell surface by activating cell type-specific receptors, such as the FcϵR activation in mast cells, 2 the oxytocin receptors in vascular endothelial cells 18 or insulin receptors in adipocytes.19,20 At present, no receptor is known to trigger IRAP recruitment in macrophages.
The hexapeptide angiotensin IV (Ang IV) is a natural inhibitor of IRAP.21,22 Ang IV is extensively studied for its memory-facilitating properties in mice and rats, however its mechanism of action remains unresolved. 23 Ang IV is also reported to protect against ischemic stroke,24,25 hyperglycemia 26 and atherosclerosis.27,28 Several research groups, including ours, therefore continue to develop synthetic Ang IV-analogues with enhanced stability and affinity for IRAP.17,29 Of particular interest, Ang IV binding to IRAP is described to activate the nuclear factor κB pathway (NF-κB) in vascular smooth muscle cells in vitro. This in turn results in an increased production of pro-inflammatory factors such as interleukin (IL)-6 and tumor necrosis factor α (TNFα).30,31
In the present study we investigated the occurrence and subcellular distribution of IRAP in mouse macrophages, as well as the regulation of its expression by pro-inflammatory mediators. We explored the function of IRAP in glucose uptake and phagocytosis, and because of the central role of the NF-κB pathway in the induction of inflammation in macrophages, 32 we investigated the activity of Ang IV and Ang IV-analogues on the expression of NF-κB regulated genes in macrophages.
Methods
Materials
Ang IV (Val-Tyr-Ile-His-Pro-Phe) was obtained from PolyPeptide Group (Strasbourg, France). AL-11 (β2hVal-Tyr-Ile-His-Pro-β3hPhe) and IVDE77 (β2hVal-Tyr-Ile-Aia-Nva-Phe) were prepared as described before.33,34 Ang IV, AL-11 and IVDE77 were used at a concentration of 1 µM, unless specified otherwise. The specific activity of tritiated [3H]IVDE77 amounted 0.90 TBq/mmol (24.4 Ci/mmol). Recombinant mouse interferon-γ (IFN-γ) (BD Biosciences, Erembodegem, Belgium) was used at 100 U/ml. Lipopolysaccharide (LPS) was obtained from Sigma (Bornem, Belgium) and used at 1 ng/ml. IL-4 (100 U/ml), IL-10 (100 U/ml) and transforming growth factor β (TGF-β) (5 ng/ml) were obtained from Peprotech (London, UK). All other reagents were of the highest grade commercially available.
Mice
IRAP wild-type (wt) and knock-out (IRAP–/–) C57BL/6 mice that were generated by Ozgene (Ozgene Ptd Ltd, Australia) as described before were bred at the animal house of the Vrije Universiteit Brussel. 35 Macrophage mannose receptor deficient (MMR–/–) mice were provided by Etienne Pays (Université Libre de Bruxelles). Signal transducers and activators of transcription deficient (STAT1–/–) mice were provided by Chantal Mathieu (Katholieke Universiteit Leuven, Belgium). Interferon regulatory factor deficient (IRF1–/–) mice were a gift of Peter Brouckaert (Universiteit Gent, Belgium). All experiments were approved by the Ethics Committee at Vrije Universiteit Brussel, Brussels, Belgium, and met the standards required by the Belgian Council for Laboratory Animal Science (BCLAS) guidelines.
Preparation and in vitro stimulation of macrophage populations
Murine naïve peritoneal macrophages (PEMs) or thioglycollate-elicited (BioMérieux, Marcy l’Etoile, France) peritoneal macrophages (thioPEMs) were obtained by rinsing the peritoneum of naïve or thioglycollate-inoculated (four days prior to cell collection) mice with PBS/10% sucrose. After 3 h culture in ME-medium (RPMI 1640 medium, 10% heat-inactivated Foetal Calf Serum (FCS), 0.03% L-glutamine, 100 mg/ml streptomycin, 100 mg/ml penicillin, 1 mM nonessential amino acids, 1 mM sodium pyruvate, and 0.02 mM 2-mercaptoethanol) in 24-well plates, non-adherent cells were washed away and the adherent peritoneal macrophages were used for analysis. To generate bone marrow-derived macrophages (BMDMs), bone marrow cells were cultured for 10 days in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 20% FCS and 30% L929 conditioned medium as a source of Macrophage-Colony Stimulating Factor (M-CSF). These macrophage types were seeded at 106 cells/ml in ME-medium and subjected to stimulation by pro-inflammatory M1-inducing mediators (IFN-γ, LPS), anti-inflammatory M2-inducing cytokines (IL-4, IL-10, TGF-β), IRAP-ligands (Ang IV, AL-11, IVDE77), or combinations thereof during 24 h unless stated otherwise.
Western blot
Total protein cell extracts were prepared as described earlier. 36 A quantity of 25 µg protein was separated on a 3–8% NuPAGE® Novex Tris-Acetate Gel (Life Technologies, Merelbeke, Belgium) and transferred onto a Hybond-C Extra nitrocellulose membrane (GE Healthcare, Diegem, Belgium). After 1 h blocking with 5% non-fat dry milk, membranes were incubated overnight at 4°C with IRAP (D7C5) XP® rabbit monoclonal antibodies (Bioké, Leiden, The Netherlands) or with mouse monoclonal to β-actin (Abcam, Cambridge, UK). After washing, membranes were incubated for 1 h with peroxidase-coupled secondary antibody. Expression was measured by electrochemiluminescence (ECL) in a Chemidoc™ XRS+ system (Bio-Rad, California, USA) and the signal was quantified using Image Lab 3.0.
Radioligand binding
General principles were described before. 37 Briefly, experiments were carried out with [3H]IVDE77 at a final concentration ranging between 1–20 nM for saturation binding and 10 nM for all other experiments. Non-specific binding was determined in the presence of 10 µM of unlabelled AL-11. The equilibrium dissociation constant (KD) and maximal number of binding sites (Bmax) were calculated by nonlinear regression analysis using GraphPad Prism 5.0 (GraphPad Software).
The distribution of intracellular versus cell surface IRAP in macrophages was investigated by the acid wash method as described earlier. 38 In brief, after 40 min incubation of thioPEMs with 10 nM [3H]IVDE77, which is not cell-permeable and hence only binds to IRAP at the cell surface, the unbound ligand was washed away. The cells were then treated with glycine buffer (pH 3.0) to disrupt and collect the cell surface bound [3H]IVDE77, corresponding to the fraction of IRAP at the cell surface. The remaining acid resistant binding thus represents the internalized [3H]IVDE77/IRAP complexes.
RNA extraction and quantitative real-time (RT) polymerase chain reaction (PCR) analysis
Total RNA extraction, cDNA synthesis and quantitative RT PCR were performed as reported earlier. 40 Primer sequences are listed in Table 1. Gene expression was always normalized using ribosomal protein S12 as housekeeping gene.
List of gene-specific primer pairs used for quantitative real-time polymerase chain reaction (RT PCR).

COX-2: cyclooxygenase-2; ICAM-1: intercellular adhesion molecule 1; iNOS: inducible nitric oxide synthase; IRAP : insulin-regulated aminopeptidase; S12 : ribosomal protein S12; TNFα: tumor-necrosis factor α.
Phagocytosis assay
IRAP–/– and wt thioPEMs were seeded in a 96-well plate and stimulated with IFN-γ (100 U/ml) and LPS (100 ng/ml). Yellow-green fluorescent latex microspheres (Polysciences, Eppelheim, Germany) were then added (diluted 1:5000) and incubated for 2 h, 24 h or 48 h at 37°C. Cells were washed three times with PBS before fluorescence of the phagocytosed particles was measured at an excitation wavelength of 440 nm and an emission of 486 nm in a TECAN® M200 spectrophotometer (Tecan Group Ltd., Mechelen, Belgium).
Flow cytometry
ThioPEMs from wt, IRAP–/– and MMR–/– mice were cultured for 24 h either with or without IL-4 (100 U/ml). Cells were then incubated for 20 min at 4°C, scraped and brought to concentration (106 cells/ml) in Hank’s Balanced Salt Solution (HBSS) supplemented with 0.5% fetal calf serum and 2 mM ethylenediaminetetraacetic acid (EDTA). Anti-FcγR antibody (2.4G2, BD Biosciences) was added to the cells for 20 min at 4°C, followed by staining (1 µg/106 cells) with anti-CD11b/PE-Cy7 (M1/70, BD Biosciences), anti-F4/80/FITC (CI: A3-1, Serotec) and anti-CD206/PE (MR5D3, Serotec) or the corresponding PE-conjugated isotype control (R35-95, BD Pharmingen). Dead cells were excluded via 7-amino-actinomycin D (7-AAD) (BD Biosciences). Data were acquired on a FACS Canto II (BD Biosciences) and analyzed by FlowJo (Tree Star).
DQ-ovalbumin (DQ-OVA, Molecular Probes) processing was performed as described before. 41 Briefly, IRAP–/– and wt thioPEMs were allowed to internalize and process DQ-OVA for 15 min at 4°C (control) or at 37°C. Free DQ-OVA was subsequently washed away and thioPEMs were allowed to process internalized DQ-OVA further for 15, 30 or 60 min. Following each time interval, cells were chilled, blocked with anti-FcγR antibody and stained with anti-CD11b/PE-Cy7 and anti-F4/80/PE. DQ-OVA fluorescence was measured via Fluorescence Activated Cell Sorter (FACS).
Cytokine quantification
Secreted TNFα levels from IRAP–/– and wt thioPEMs cultures were quantified using sandwich ELISA following manufacturer’s instructions (DY410, R&D Systems).
Glucose uptake assay
IRAP–/– and wt thioPEMs were seeded in 24-well plates and used either unstimulated or stimulated for 24 h with IFN-γ or LPS (100 ng/ml). Cells were washed three times with PBS and incubated for 20 min at 37°C in buffer alone or with 20 µM cytochalasin B (Sigma, Bornem, Belgium) to block glucose transport 39 . 2-deoxy-D-[3H]glucose (Perkin Elmer, Zaventem, Belgium) was then added at a final concentration of 1 mM (corresponding to 1 µCi/ml) and incubated for 10 min at 37°C. Uptake of 2-deoxy-D-[3H]glucose was stopped by washing the cells three times with PBS on ice. Cells were then lysed with 1 M NaOH and the accumulated 2-deoxy-D-[3H]glucose was measured after adding 3 ml of scintillation liquid (Optiphase Hisafe) using a β-counter (Perkin-Elmer, Zaventem, Belgium). Data refer to specific 2-deoxy-D-[3H]glucose uptake, calculated by subtracting non-specific uptake in the presence of 20 μM cytochalasin B.
Statistical analysis
All experiments were performed at least three times with triplicate determinations each, unless stated otherwise. Radioligand saturation curves were obtained by analyzing the data according to a one-site model using GraphPad Prism 5.0 software (GraphPad Software). Statistics were performed by one-way analysis of variance (ANOVA), and when p<0.005, Dunnett’s post-hoc test was performed to compare each condition with the control. *p<0.05; **p<0.005; ***p<0.001; ns: not significant.
Results
Different mouse macrophage types express similar IRAP protein levels
The IRAP protein levels in different mouse macrophage types (naïve PEMs, thioPEMs and BMDMs) were investigated by Western blotting. IRAP protein expression was comparable among all the macrophages tested and was absent in IRAP–/– macrophages, validating the IRAP-specificity of the tested antibody (Figure 1). Notably, IRAP protein levels in macrophages were twofold lower than those in differentiated 3T3-L1 adipocytes, which are known as typical IRAP-expressing cells.16,42,43

Different mouse macrophage types express similar insulin-regulated aminopeptidase (IRAP) protein levels. IRAP protein content was compared between mouse naïve peritoneal macrophages (PEMs) (lane 1), thioglycollate-elicited peritoneal macrophages (thioPEMs) (lane 2) and bone marrow-derived macrophages (BMDMs) (lane 3). Differentiated 3T3-L1 adipocytes (3T3-L1), a typical IRAP-expressing cell line, served as a positive control (lane 5). ThioPEMs from IRAP–/– mice served as a negative control (lane 4). A representative of two experiments is shown here.
IRAP is mainly located intracellularly in resting macrophages
To investigate the presence of IRAP at the cell surface of live macrophages, saturation binding experiments were performed in vitro with the Ang IV-analogue [3H]IVDE77. 17 Specific binding of [3H]IVDE77 to wt thioPEMs was clearly detectable, and the binding parameters were determined by non-linear regression (KD 6.71 nM±0.64 nM; Bmax 0.27 pmol/106 cells±0.01 pmol/106 cells) (Figure 2(a)). ThioPEMs derived from IRAP–/– mice showed no specific binding, confirming that the measured radioactivity solely resulted from [3H]IVDE77 binding to IRAP.
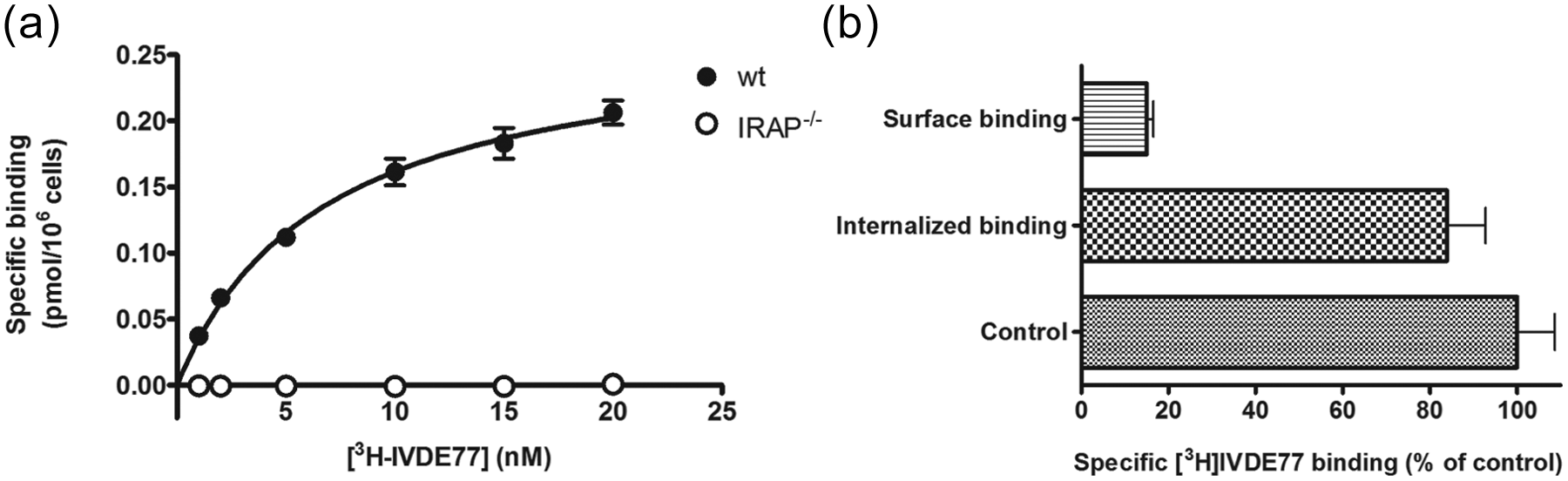
Insulin-regulated aminopeptidase (IRAP) is mainly located intracellularly in resting macrophages. (a) Saturation binding to wild-type (wt) and IRAP–/– thioglycollate-elicited peritoneal macrophages (thioPEMs) was carried out with [3H]IVDE77 at a final concentration ranging between 1–20 nM. Data refer to specific binding (expressed as pmol/106 cells), calculated by subtracting non-specific binding in the presence of 10 μM AL-11 from total binding. A representative of two independent experiments is illustrated. (b) The distribution of intracellular versus cell surface IRAP was investigated by the acid wash method. Glycine buffer treatment (pH 3.0) disrupted only the cell membrane bound [3H]IVDE77, corresponding to the fraction of IRAP on the cell surface. The remaining acid resistant binding represents the internalized [3H]IVDE77/IRAP complexes. Values are expressed as % binding compared to their corresponding untreated control condition.
Next we investigated the distribution of intracellular versus cell surface IRAP in wt macrophages by the acid wash method. 38 Under basal conditions, only 15% of [3H]IVDE77/IRAP complexes were located at the cell surface of thioPEMs, while the remaining bound [3H]IVDE77 was detected intracellularly (Figure 2(b)). These results indicate that the translocation of IRAP in macrophages is a dynamic process, in accordance with earlier observations in Chinese hamster ovary cells. 37
IRAP is higher expressed in M1-activated macrophages
Macrophages are very modular cells that are especially responsive to their environment.32,45,46 We therefore characterized the expression and regulation of IRAP in macrophages, to determine in which macrophage activation state (pro-inflammatory M1 or anti-inflammatory M2) IRAP may be more prominent.
Compared to the control condition, expression of IRAP mRNA rose almost threefold after 24 h stimulation with IFN-γ (2.68±0.62) (Figure 3). Using STAT1–/– and IRF1–/– macrophages, we demonstrated that the IFN-γ-mediated IRAP mRNA induction entirely depends on STAT1, but is independent of IRF1, which is a STAT1-dependent transcription factor responsible for the expression of ‘second wave’ genes (Figure 4). Besides IFN-γ, IRAP mRNA was significantly induced by LPS (4.31±1.73). Combined stimulations of thioPEMs with IFN-γ and LPS similarly enhanced IRAP expression (3.72±1.70). On the other hand, anti-inflammatory cytokines (IL-4, IL-10, TGF-β) did not significantly influence IRAP expression. These data indicate that IRAP mRNA is more expressed by pro-inflammatory M1-type macrophages.

Interferon-γ (IFN-γ) and lipopolysaccharide (LPS) upregulate insulin-regulated aminopeptidase (IRAP) mRNA expression in thioglycollate-elicited peritoneal macrophages (thioPEMs). ThioPEMs were treated with the respective cytokines and LPS for 24 h and IRAP mRNA expression was assessed via real-time polymerase chain reaction (PCR). Fold induction of IRAP gene expression is plotted relative to the expression level in untreated macrophages (=1). Each dot represents an individual experiment.

Interferon-γ (IFN-γ)-mediated insulin-regulated aminopeptidase (IRAP) mRNA expression is signal transducers and activators of transcription (STAT1)-dependent, but interferon regulatory factor ( IRF1)-independent. STAT1-deficient (STAT1–/–) and IRF1-deficient (IRF1–/–) thioglycollate-elicited peritoneal macrophages (thioPEMs) were treated with IFN-γ for 24 h and IRAP mRNA expression was assessed via real-time polymerase chain reaction (PCR). Fold induction of IRAP gene expression is plotted relative to the expression level in the corresponding STAT1–/– and IRF1–/– untreated macrophages (=1). Each • represents an independent cytokine stimulation, followed by RNA-extraction and real-time PCR. **p<0.005; ns: not significant.
Next, the modulation of IRAP protein expression was investigated by Western blot. In line with the mRNA data, IFN-γ and LPS significantly increased IRAP protein expression compared to control samples (1.99±0.28 and 1.69±0.51, respectively) (Figure 5). The same expression pattern was also observed after 48 h stimulation of thioPEMs with IFN-γ or LPS (data not shown).

Interferon-γ (IFN-γ) and lipopolysaccharide (LPS) upregulate insulin-regulated aminopeptidase (IRAP) protein expression in thioglycollate-elicited peritoneal macrophages (thioPEMs). (a) Western blotting was performed on total protein extracts from thioPEMs stimulated for 24 h with IFN-γ, LPS or medium. Mean values were calculated from four independent experiments, each normalized to the protein content of β-actin in their respective lanes. A representative experiment is shown in (b). *p<0.05; **p<0.001.
IFN-γ and LPS differentially modulate IRAP-presence at the cell surface
Since we observed a significantly higher gene and protein expression of IRAP after treatment with IFN-γ or LPS, we assessed whether this upregulation translates into a higher presence of IRAP at the cell surface of thioPEMs by [3H]IVDE77 binding.
IFN-γ indeed increased [3H]IVDE77 binding, steadily over time after 2 h (114%±3% of control), 24 h (171%±23%) or 48 h (179%±23%) (Figure 6). Strikingly, a different pattern was seen after LPS-treatment. Initially, LPS induced a higher amount of IRAP at the cell surface after 2 h (137%±19%), but then decreased [3H]IVDE77 binding to control levels after 24 h (99%±19%) and even lower after 48 h (74%±4%). This decrease of [3H]IVDE77 binding could not be explained by changes in cell viability as checked by Trypan blue staining. Combined stimulations with IFN-γ and LPS caused effects similar to LPS alone (147%±14% after 2 h, 119%±12% after 24 h, 75%±6% after 48 h).

Interferon-γ (IFN-γ) and lipopolysaccharide (LPS) differentially modulate insulin-regulated aminopeptidase (IRAP) presence at the cell surface. Thioglycollate-elicited peritoneal macrophages (thioPEMs) were pre-incubated for the indicated time periods with medium, IFN-γ, LPS or IFN-γ and LPS combined. Binding was then initiated with [3H]IVDE77 (10 nM, final concentration) and further incubated for 30 min at 37°C. Values are expressed as % binding compared to their corresponding untreated control condition. *p<0.05, ***p<0.001.
IRAP is recruited to the cell surface during phagocytosis
Recombinant soluble IRAP was reported to augment the phagocytic uptake of latex beads by IFN-γ/LPS-stimulated RAW264 macrophages. 47 We therefore investigated whether latex beads could recruit IRAP to the cell surface, and whether IRAP-deficiency would decrease phagocytosis in thioPEMs. Incubating wt thioPEMs for 2 h with latex beads significantly increased subsequent [3H]IVDE77 binding (147%±17%) (Figure 7(a)). Remarkably, incubating the cells with latex beads for 24 h or 48 h did not show increased IRAP surface expression, indicating that IRAP recruitment to the cell surface was transient.

Insulin-regulated aminopeptidase (IRAP) is recruited at the cell surface during phagocytosis. (a) Thioglycollate-elicited peritoneal macrophages (thioPEMs) were pre-incubated for the indicated time periods with fluorescent latex beads. Binding was then initiated with [3H]IVDE77 (10 nM, final concentration) and further incubated for 30 min at 37°C. 0h(*) represents the condition of simultaneous addition of latex beads and [3H]IVDE77. Values are expressed as % binding compared to their corresponding untreated control condition; (b) IRAP–/– and wild-type (wt) thioPEMs were incubated for the indicated time periods with fluorescent latex beads together with medium or interferon-γ (IFN-γ) plus lipopolysaccharide (LPS). Cells were washed and the ingested fluorescence was measured; (c) wt thioPEMs were pre-incubated for 1 h with 1 µM IVDE77 to block the enzymatic activity of IRAP. Cells were then incubated with fluorescent latex beads for the indicated time periods, and the ingested fluorescence was measured. ***p<0.001, ns: not significant.
To assess the role of IRAP recruitment in phagocytosis, the fluorescence of ingested latex beads was measured at different time points in wt and IRAP–/– thioPEMs. Basal uptake increased gradually over time, in a similar fashion in both wt and IRAP–/– thioPEMs (Figure 7(b)). Phagocytic uptake was more pronounced after treatment with IFN-γ/LPS, but no significant difference between wt and IRAP–/– thioPEMs was observed. Pre-treating wt thioPEMs for 1 h with 1 µM IVDE77 to block the enzymatic activity for IRAP also did not influence phagocytosis of latex beads (Figure 7(c)).
IRAP-deficiency does not alter macrophage mannose receptor (MMR)-mediated endocytosis in macrophages
Besides phagocytosis of large particles, macrophages also ingest molecules via receptor-mediated endocytosis. In this context, MMR-mediated endocytosis is of particular interest, since IRAP has been reported to co-localize with MMR.3,48 Therefore, we first assessed whether IRAP-deficiency could influence the distribution of MMR. No difference in MMR-positive cells was observed by flow cytometry between IRAP–/– and wt thioPEMs (Figure 8(a)). IL-4 stimulation also increased MMR expression similarly in both groups. Additionally, we tested whether MMR-deficiency would alter the distribution of IRAP. No difference in [3H]IVDE77 binding to IRAP was observed between wt and MMR–/– thioPEMs (Bmax,wt: 0.26 pmol/106 cells±0.01 pmol/106 cells; Bmax,MMR-KO: 0.31 pmol/106 cells±0.10 pmol/106 cells) (Figure 8(b)). These data indicate that although IRAP and MMR may co-localize, their distribution is regulated independently of each other in macrophages.

Insulin-regulated aminopeptidase (IRAP)-deficiency does not alter macrophage mannose receptor (MMR) distribution in macrophages. (a) Untreated or interleukin-4 (IL-4) treated IRAP–/– and wild-type (wt) thioglycollate-elicited peritoneal macrophages (thioPEMs) were analyzed for MMR expression by flow cytometry. Histograms represent isotype control (shaded) and surface MMR–specific (black line) staining within the gated population. The percentages given are the normalized median fluorescence intensities (nMFIs), calculated as nMFI=(MFIsample – MFIisotype)/MFIsample. MMR–/– thioPEMs served as a negative control. A representative of two experiments is shown here. (b) IRAP-distribution between wt and MMR–/– thioPEMs was analyzed by [3H]IVDE77 saturation binding as described earlier. A representative of two experiments is shown here.
OVA uptake by macrophages is MMR-dependent. 49 Hence, to test whether IRAP influences MMR functionality, the kinetics of DQ-OVA uptake and degradation by wt versus IRAP–/– macrophages was assessed using flow cytometry. However, no differences between both macrophage types could be observed (Figure 9).

Insulin-regulated aminopeptidase (IRAP) does not influence the rate of macrophage mannose receptor (MMR)-mediated antigen uptake and degradation. IRAP–/– and wild-type (wt) thioglycollate-elicited peritoneal macrophages (thioPEMs) were allowed to internalize and process DQ-ovalbumin (DQ-OVA) for 15 min at 4°C (control) or at 37°C. Free DQ-OVA was subsequently washed away and thioPEMs were allowed to process internalized DQ-OVA further for 15, 30 or 60 min. Following each time interval, cells were surface labeled and DQ-OVA fluorescence in IRAP–/– and wt thioPEMs was measured via FACS. Values are shown as % of DQ-OVA positive cells in relation to the total live gated thioPEMs.
IRAP-deficiency does not influence glucose uptake in macrophages
In fat and muscle cells, IRAP is co-localized with the glucose transporter GLUT4 and is as such involved in insulin-mediated glucose uptake.13,50 In addition, GLUT4 might contribute to meeting the enhanced energy needs of activated monocytes. 51
We therefore investigated whether IRAP plays a role in the glucose uptake of macrophages. No difference was observed in the uptake of 2-deoxy-D-[3H]glucose between wt and IRAP–/– thioPEMs under basal conditions (Figure 10(a)). Glucose uptake strongly increased (approximately 50% higher compared to control) in both wt and IRAP–/– thioPEMs after 24 h activation with IFN-γ or LPS (Figure 10(b)). These data indicate that IRAP may not be implicated in glucose uptake by macrophages, both under basal and inflammatory conditions.

Insulin-regulated aminopeptidase (IRAP)-deficiency does not influence glucose uptake in thioglycollate-elicited peritoneal macrophages (thioPEMs). (a) IRAP–/– and wild-type (wt) thioPEMs were incubated with 1 mM 2-deoxy-D-[3H]glucose (corresponding to 1 µCi/ml) for 10 min at 37°C, followed by washing on ice. Data refer to specific 2-deoxy-D-[3H]glucose uptake, calculated by subtracting non-specific uptake in the presence of 20 μM cytochalasin B. Each point represents an individual experiment with triplicate samples. (b) IRAP–/– and wt thioPEMs were stimulated for 24 h with medium, interferon-γ (IFN-γ) or lipopolysaccharide (LPS). Cells were washed three times with PBS, and incubated with 1 mM 2-deoxy-D-[3H]glucose for 10 min at 37°C. Values are expressed as % uptake compared to the corresponding untreated control condition, ns: not significant.
IRAP ligands do not modulate NF-κB regulated gene expression in macrophages
In view of the potential impact of IRAP binding on the NF-κB pathway,30,31 we investigated the modulatory activity of Ang IV and the stable Ang IV-analogue AL-11 33 on the expression of NF-κB regulated genes (inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), tumor-necrosis factor α (TNFα), intercellular adhesion molecule 1 (ICAM-1)) in macrophages by quantitative real-time PCR (Table 1). None of these genes were induced by Ang IV or AL-11, suggesting that IRAP binding does not directly stimulate NF-κB signaling in macrophages (Figure 11).

Insulin-regulated aminopeptidase (IRAP) ligands do not induce the expression of nuclear factor kB pathway (NF-κB) regulated genes in macrophages. Wild-type (wt) thioglycollate-elicited peritoneal macrophages (thioPEMs) were subjected to 24 h stimulation with 1 µM angiotensin IV (Ang IV) or AL-11 and the expression of : inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), tumor-necrosis factor α (TNFα) and intercellular adhesion molecule 1 (ICAM-1) was analyzed by quantitative real-time polymerase chain reaction (PCR). Each • represents an independent stimulation with Ang IV or AL-11, followed by RNA-extraction and real-time PCR.
Next, thioPEMs were subjected to LPS in combination with Ang IV or AL-11 to analyze the potential modulating effect of Ang IV/AL-11 in an inflammatory setting.52,53 Ang IV did not significantly affect the LPS-induced expression of NF-κB regulated genes (iNOS, COX-2, TNFα, ICAM-1) (Figure 12(a)). In the case of treatment by AL-11, a reduction in the LPS-induced expression of iNOS and TNFα, only reaching significance in the case of TNFα (54.9%±26.7%), could be observed in macrophages (Figure 12(b)). However, AL-11 did not significantly influence the secretion of TNFα by LPS-stimulated macrophages, illustrating that IRAP binding does not have a major impact on the inflammatory status of these cells (Figure 12(c)).

Insulin-regulated aminopeptidase (IRAP) ligands do not significantly modulate lipopolysaccharide (LPS)-induced inflammation in macrophages. Wild-type (wt) thioglycollate-elicited peritoneal macrophages (thioPEMs) were subjected to 24 h stimulation with LPS in combination with 1 µM angiotensin IV (Ang IV) (a) or AL-11 (b), after which the LPS-induced expression of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), tumor-necrosis factor α (TNFα) and intercellular adhesion molecule 1 (ICAM-1) was analyzed by quantitative real-time polymerase chain reaction (PCR). Values are expressed as % mRNA expression compared to the LPS alone condition. Each • represents an independent cytokine stimulation, followed by RNA-extraction and real-time PCR. (c) Influence of Ang IV and AL-11 on TNFα secretion from thioPEMs. The supernatant of the cultured thioPEMs as described above was collected and tested via TNFα ELISA. Each point represents an individual experiment with triplicate samples. **p<0.005; ns: not significant.
Discussion
For the first time, the presence and regulation of IRAP was investigated in mouse macrophages. Total IRAP protein expression was comparable between naïve PEMs, thioPEMs and BMDMs (Figure 1). These data are in line with the IRAP protein levels reported in dendritic cells, which are similar between dendritic cell subtypes.3,54 The presence of IRAP at the cell surface of macrophages was shown by specific binding of the Ang IV-analogue [3H]IVDE77. No binding was observed in IRAP–/– macrophages, indicating that no alternative binding sites nor specific uptake mechanisms of [3H]IVDE77 take place. In all cell types examined so far, IRAP localizes mainly in the perinuclear region in specialized secretory vesicles.2,16,17 By use of the acid wash method, we confirmed that only 15% of the total IRAP pool was present at the cell surface of thioPEMs, while the remaining [3H]IVDE77/IRAP complexes were found intracellularly (Figure 2).
The transcriptional expression of IRAP was examined in thioPEMs after 24 h treatment with pro-/anti-inflammatory mediators. We observed that IFN-γ induced a threefold upregulation of IRAP mRNA expression, which was mediated by the transcription factor STAT1 but not by IRF1 (Figures 3 and 4). This translated into a corresponding higher IRAP protein content and [3H]IVDE77 binding in thioPEMs (Figures 5 and 6). These IFN-γ mediated effects are very similar to the data of Gabrilovac and coworkers, 8 which showed that IFN-γ induces a twofold increase in AP-N mRNA and surface expression in the macrophage cell line J774.
LPS-treatment also increased IRAP mRNA and protein expression in thioPEMs. Minimally modified low-density lipoprotein (mmLDL), which as LPS binds to Toll-like receptor 4, 55 was reported to increase IRAP-expression in foam cells under high glucose and insulin conditions. 56 Together with our observation that anti-inflammatory cytokines (IL-4, IL-10, TGF-β) did not induce IRAP-expression, these data demonstrate that IRAP is more expressed in pro-inflammatory M1-activated macrophages, and suggest it may play a role in cytotoxic or inflammation-associated functions.
A surprising pattern in [3H]IVDE77 binding was observed in LPS-treated thioPEMs. While a relatively short pre-incubation of 2 h with LPS augmented [3H]IVDE77 binding, it subsequently declined after longer time periods (24 h and 48 h) (Figure 6). A possible explanation for the rapid increase in [3H]IVDE77 binding is that LPS-treatment might induce recruitment of IRAP-vesicles to the cell surface of macrophages. Trafficking of IRAP-vesicles is known to be triggered by different stimuli and is cell type-dependent. 3 For example, IRAP is quickly recruited to the cell surface of mast cells upon FcϵR-activation with opsonized antigen, 2 or by oxytocin binding to its receptor in human vascular endothelial cells. 18 In this context, we also observed a transient increase in [3H]IVDE77 binding to thioPEMs after incubation with latex particles (Figure 7). Further work is clearly required to identify the stimuli and the involved signaling pathways responsible for IRAP-trafficking in macrophages.
Increasing evidence points towards the importance of IRAP in an immunological context. For instance, IRAP is reported to be crucial for N-terminal trimming of exogenous antigens that are directed for cross-presentation on MHC-I in dendritic cells.48,54 Also, addition of recombinant IRAP in the culture medium augments phagocytosis of latex beads in IFN-γ/LPS stimulated RAW264-macrophages. 47 While we observed that fluorescent latex beads transiently recruited IRAP to the cell surface, phagocytosis of these fluorescent beads was however similar between wt and IRAP–/– thioPEMs, suggesting that the enzymatic activity of IRAP is not required for the ingestion of particles. Given that the N-terminal cytosolic tail of IRAP interacts with several proteins implicated in protein sorting, vesicle formation and cytoskeleton remodeling, we speculate that recruitment of IRAP-containing vesicles may function as a shuttle, delivering important yet undefined proteins at the cell surface to respond against LPS or exogenous particles.3,20,57–60
The C-type lectin receptor MMR is responsible for receptor-mediated endocytosis of antigens containing high-mannose structures (such as OVA). 61 MMR has been shown to be associated with AP-N in the macrophage cell line J774, 8 and in dendritic cells, MMR colocalizes up to 75% with IRAP. 48 In this context, our data indicate however that no interdependency exists between the cell surface distribution of IRAP and MMR in macrophages (Figure 8). No difference was also observed in the MMR-mediated uptake and processing of DQ-OVA between IRAP–/– and wt thioPEMs (Figure 9).
Macrophages involved in an immune response demonstrate an increased glucose demand to meet the energy needs for activation.62,63 In this context, uptake of glucose represents the rate-limiting step.64,65 Hence, a rise in translocation of glucose transporters (including GLUT4) was reported in human monocytes after activation by LPS. 51 Since IRAP affects GLUT4-trafficking, 57 we investigated whether IRAP-deficiency would disturb glucose uptake in macrophages. However, no difference in glucose uptake was observed between wt and IRAP–/– thioPEMs, either under resting, IFN-γ or LPS-activated conditions (Figure 10). IRAP may thus not be implicated in glucose uptake, as supported by the observation that glucose uptake increased by about 50% after 24 h LPS treatment, while no increase in [3H]IVDE77 binding was seen under the same conditions. These data do not exclude the contribution of GLUT4 in glucose uptake during inflammation, nor the existence of potential compensation mechanisms upregulating other glucose transporters in IRAP–/– thioPEMs.
A number of papers report activation of signaling pathways upon binding of Ang IV to IRAP, implying a receptor function for IRAP.26,66 –69 Of note, Ang IV activates the inflammatory NF-κB pathway in vascular smooth muscle cells, which leads to the production of pro-inflammatory factors such as IL-6, TNFα and ICAM-1. 30 We therefore investigated any potential modulatory effect of Ang IV (and the Ang IV-analogue AL-11) on the NF-κB pathway in macrophages. Expression of the NF-κB regulated genes iNOS, COX-2, TNFα or ICAM-1 was not induced by Ang IV or AL-11 (Figure 11). On the other hand, an anti-inflammatory trend could be observed, in particular the LPS-induced expression of TNFα was significantly reduced in AL-11 treated macrophages. Secreted TNFα levels were however unaffected by Ang IV or AL-11. In the current debate of IRAP functioning as a receptor, 70 these data tend towards Ang IV as an anti-inflammatory mediator.
Altogether, it is clear that IRAP plays different roles depending on the cell type and extracellular environment. In the case of macrophages, the role of (Ang IV binding to) IRAP has just started to be unraveled. We showed here that IRAP is higher expressed in pro-inflammatory M1-activated macrophages, and that its presence at the cell surface varies upon exposure to IFN-γ, LPS or exogenous particles. A further understanding of the regulation and translocation of IRAP in macrophages by use of synthetic Ang IV-analogues will contribute substantially to elucidating the function(s) of IRAP in an immunological context.
Footnotes
Acknowledgements
The authors thank Ella Omasta, Marie-Therese Detobel, Maria Slazak, Nadia Abou and Eddy Vercauteren for technical and administrative assistance. They also acknowledge Marco Benatar for proofreading this manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Funding
This work was supported by the Research Council of the Vrije Universiteit Brussel (GOA-2007); the Agency for Innovation by Science and Technology in Flanders (IWT 093395) and the Geneeskundige Stichting Koningin Elisabeth.