
Abstract
Hypothesis/introduction:
The relationship between salt intake, blood pressure and RAAS activation is still controversial, being that both high- and low-salt intakes are associated with cardiovascular events in a J-shaped curve pattern. We hypothesized that different patterns of RAAS response to dietary salt intake among hypertensives could be identified, while vascular damage would be related to high-salt intake plus absence of expected RAAS inhibition.
Objective:
We aim to assess the relationship between sodium intake, RAAS and vascular stiffness in hypertension.
Materials and methods:
We screened 681 hypertensive patients for urinary/plasma electrolytes, renin, aldosterone and pulse wave velocity (PWV) under their usual salt intake level.
Results:
After applying exclusion criteria, an inverse relation between urinary sodium and RAAS was observed in the 300 remaining subjects. Additionally, four types of response were identified: 1) Low (L) sodium (S)-Low RAAS, 2) LS-High (H) SRAAS, 3) HS-Low RAAS, 4) HS-High RAAS. We found no differences in age/BP among groups, but type 4 response individuals included more females and a higher pulse wave velocity.
Conclusions:
We showed a) an inverse salt-RAAS relation, b) an association between HS plus high RAAS with increased PWV that could identify a higher-risk hypertensive condition.
Introduction
In a recent revision, Kotchen et al. 1 describe the relationship between salt intake, blood pressure, and vascular stiffness as a “delicate balance.” A large number of epidemiological studies have established a generally curvilinear relationship between dietary salt intake, inferred from urinary sodium excretion or dietary history, and the occurrence of hypertension.2–5 But at the same time, there are also a limited number of observational studies that demonstrate an increased prevalence of cardiovascular disease associated with low-salt intake. 6 O’Donnell et al. 7 suggest that both high- and low-sodium intake were associated with increased cardiovascular events in a J-shaped curve. The exact pathway of the salt-mediated cardiovascular damage is not well established. Preclinical evidence supports a link between high-salt intake, mineralocorticoid level, and the development of rapid vascular damage in essential hypertension. In animal models of hypertension, aldosterone excess in addition to high dietary salt intake contributes directly to cardiovascular impact, but this deleterious effect is reduced or even prevented by low-salt ingestion.6,8,9 However, limited data are available to determine whether such an interaction between aldosterone and dietary salt, which would promote cardiovascular damage, occurs in humans. An approximation of this determination was provided by Pimenta and Calhoun, who demonstrated that the association between aldosterone excess and high dietary salt promotes an increase in urinary protein excretion among hypertensive patients. 10 It is generally accepted that under normal renal and vascular conditions, the response to high-salt (HS) intake involves the inhibition of renin and aldosterone secretions (RAAS), whereas a low-sodium intake stimulates them. 11 Despite this close homeostatic relation, another mechanism for the rapid and chronic elimination of HS loads depends on a suitable response of pressure-induced natriuresis, and a complex network of biomolecular and vascular mediators. 12 The mechanism by which an HS diet may activate RAAS is not completely elucidated, and is probably related to the depletion of circulating angiotensin II, which in turn upregulates AT1 receptors. 13 Interestingly, it was also demonstrated that angiotensin II type 1 receptor (AT1R) mRNA and/or protein increased in the hearts of normal rats during HS intake, suggesting that the cardiac RAAS may be activated independently of arterial pressure or circulating RAAS status. 14
As described by Schlaich and others, HS intake not only affects blood pressure but also promotes cardiac and vascular fibrosis.14–17 Substantial evidence actually shows that aldosterone promotes adverse functional and structural alterations of the vessel walls independently from blood pressure mainly by changes in the elastin, calcium, and collagen densities and turnover.18–20
A higher RAAS activation associated with the individual sodium intake pattern would probably be related to vascular stiffness and increased hypertensive cardiovascular risk. We hypothesized that in a daily clinical setting, the analysis of RAAS levels in response to dietary salt intake in hypertensive patients would allow us to identify different pathophysiologic profiles probably related to the status of their individual salt regulatory mechanism. Moreover, we also supposed that the coexistence of high-salt intake and a high RAAS status are both necessary conditions for the development of rapid vascular damage in hypertensive patients.
Based on this, the aims of our study were to assess whether different sodium loads are associated with diverse patterns of RAAS activation, and to identify the relation between sodium intake, RAAS activation patterns, and vascular stiffness.
Materials and methods
Study individuals
Patients with essential hypertension aged 30 to 70 years were invited to participate while they spontaneously attended or were referred by their primary care physician to the Centre of Hypertension at the Hospital Universitario Austral, from January 2011 to October 2012. To be included in the study, they had to be able to complete a 14- to 21-day washout period of antihypertensive drug treatment, had to have normal renal function, and had to have any known factor that could modify the renin and aldosterone system (RAAS). Patients were instructed to follow their usual sodium diet during that period, but no control was planned in the study protocol in order to obtain data representative of the individual’s real life with a broad spectrum of salt intake among participants.
Exclusion criteria
The presence of any of the following symptoms was considered as exclusion criteria and the patient was not included in the study: abnormal renal function, volume or electrolyte alterations, diabetes, history of renal disease, ischemic heart disease, stroke, loss of data, counterindication for the drug washout, use of corticoids or nonsteroidal anti-inflammatory drugs during the study.
Blood pressure (BP) measurement
According to the European Society of Hypertension guidelines, 21 office BP was measured with a validated semiautomatic oscillometric device (Omron HEM-705) the morning of the study.
Determination of aldosterone (Aldo) and plasma renin activity (PRA) status
Peripheric blood samples to determine PRA and Aldo were obtained after overnight (12 hours) fasting and a 30-minute rest period (RIA) in the supine position in a quiet room between 7 and 8 a.m. (RIA). Low/high values were defined using normal reference values of PRA < or > 2.2 ng/ml/h; Aldo < or > 160 mg/ml. The same sample was used for routine determinations, serum electrolytes, and creatinine. Spontaneously voided urine was collected over a 24-hour period and analyzed for Na+, K+, Cl–, and creatinine. To avoid possible pharmacological interference with RAAS, no antihypertensive drugs were allowed during a 21-day period prior to the determinations (washout period). If necessary, BP reduction was obtained using doxazocine or verapamilo.
Plasma and red blood cells were separated by centrifugation, and PRA (ng Angiotensin II/ml/h) and Aldo levels (ng/ml) were determined.
As we previously reported 22 our intra-assay and inter-assay coefficients of variation are 5.4 and 9.1%, respectively, for PRA; and 7.9 and 9.6%, respectively, for Aldo.
Salt intake analysis and group classification
Salt dietary load was estimated by analysis of 24-hour sodium excretion. We used the World Health Organization (WHO) salt intake recommendation for hypertensive patients, which must be < 60 mmol/l or 90 mmol/24 hours and we classified subjects in two groups; those within this value were considered as low sodium and those above it as high sodium. 23
Pulse wave velocity (PWV) determination
PWV was synchronically calculated between the left carotid artery and the left femoral artery. The pulse waveform was recorded at each site with tonometry and the time delay between the arrival at the two sites was calculated. Measurement of distance of the body surface between the femoral and carotid artery estimated the distance traveled. PWV is shown as distance/time (m/s) using Hemodyn 4M/TM.
Statistical analysis
Statistical analysis was carried out using a validated statistic software package (MedCalc, v 11.3.0.0). Continuous variables with normal distribution were expressed as mean ± SD, and variables with skewed distribution (NaUr, Aldo, and PRA) were described through the median and the respective interquartile ranges (25%−75%). Analysis of the differences in age, sex, BP, NaUr, PWV, and RAAS status according to the different patterns of sodium intake/RAAS responses were performed by analysis of variance (ANOVA) one-way test in normal-distributed variables. Variables with skewed distribution were analyzed through a Kruskall Wallis test. A linear correlation between NaUr and Aldo was performed (Spearman test). A p value < 0.05 was considered statistically significant.
Results
A total of 685 hypertensive patients were screened, and after applying exclusion criteria a final population of 300 patients was included in the study. Table 1 shows sex, age, systolic and diastolic BP, time elapsed from diagnosis of hypertension, and the number and type of antihypertensive drugs taken before the washout period of each patient. On the day of sample collection and without any antihypertensive pharmacological treatment, BP was higher than 140 mmHg or 90 mmHg, systolic and diastolic respectively, in 62% of the patients; while 24% had normal BP values with a rescue antihypertensive drug treatment that had been indicated at least one week before, and 14% were normotensive without any drug.
Population baseline characteristics.

ACE: angiotensin converting enzyme; AT1: angiotensin I.
An inverse relation between sodium output and RAAS was clearly demonstrated when results for the whole population were analyzed as shown in Figure 1 (sodium-aldosterone) and Figure 2 (sodium-renin).
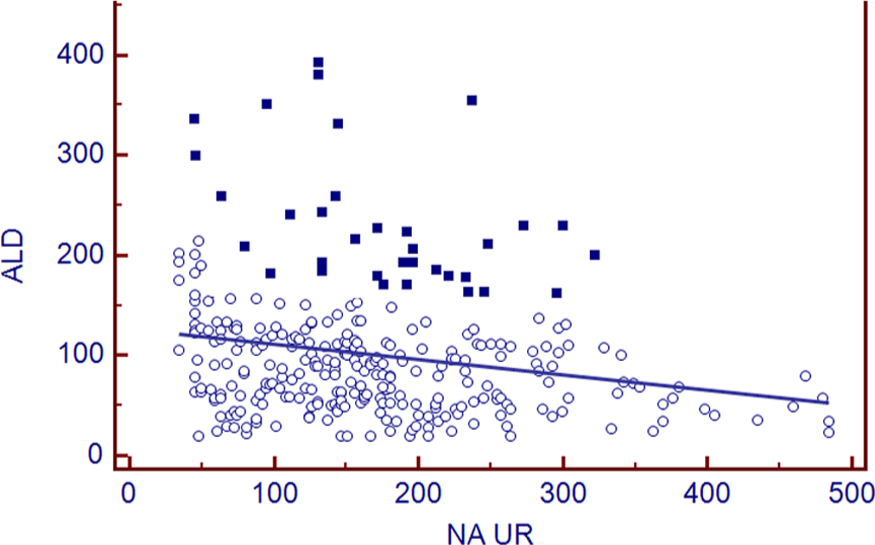
Relationship between plasma aldosterone and urinary sodium output (24 hours) in essential hypertensive patients demonstrated a linear correlation that represents almost 90% of the entire population (circle dots). A small group of patients show a nonlinear relationship whereas high sodium intake values were associated with high aldosterone values (square dots).

Relationship between plasma renin activity and urinary sodium output (24 hours) in essential hypertensive patients.
Individual analysis showed that a low-sodium intake was associated with two different RAAS patterns as follows: a) low sodium-low RAAS and, b) low sodium-high RAAS. A similar response was observed among those with high-sodium intake; c) High-sodium-low RAAS, and d) high-sodium-high RAAS.
These results supported the classification of the population on the basis of the type of response observed as shown in Table 2.
Relation between RAAS and dietary sodium intake among essential hypertensives.

RAAS: Renin Angiotensin Aldosterone System; SBP: systolic blood pressure; DBP: diastolic blood pressure; Sodium: sodium intake inferred from 24-hour urinary output; Aldosterone: plasma aldosterone; PRA: plasma renin activity; H: high; L: low. Reference values: High/Low sodium: >/< 90 mEq/l; High/Low Aldosterone: >/<160 mg/ml; High/Low PRA: >/<2.2ng/ml/h; NS: not significant.
Distribution by age and blood pressure were similar between the four groups. The high-sodium-high RAAS group had a significantly higher number of women. PWV was also found to be increased in the high-sodium-high RAAS group.
Aldosterone to renin ratio was < 20 in all groups, a condition that was especially evaluated among those with high aldosterone not suppressed by salt, which can normally diagnose primary hyperaldosteronism; however, they also had high (not low) renin.
Discussion
There are two major findings in this study: First, we verified an inverse relation between salt intake and RAAS activation, and second, we also found that in a small group of patients HS intake was related to a physiologically unexpected increase in RAAS activation that was accompanied by accelerated vascular damage.
Our first finding is coincident with results communicated in 1998 by Graudal et al., 11 whose extensive meta-analysis explored the effects of sodium restriction on blood pressure, renin, aldosterone, and catecholamines. Their data showed that those patients with a sodium intake reduction of 40 to 100 mmol/24 hours presented significant increases in plasma renin and aldosterone by a factor of 1.8 to 2.5 and two-fold, respectively. This response did not differ between normotensive and hypertensive patients, whereas the noradrenaline response was much stronger in hypertensive subjects. The inverse relation between salt and RAAS makes it difficult to explain how a low-salt diet could promote cardiovascular health when circulating angiotensin II levels are increased. Some authors speculated about the existence of reduced tissue responsiveness to angiotensin II, but it can be argued that aldosterone is also increased in this condition. Another possible condition could be related to modifications of AT1 receptor vascular expression in response to changes in dietary salt intake, but that has not been clinically explored. 12
Our second finding shows that activation of RAAS response to salt load is not similar among all hypertensive patients. The unexpected pattern of high RAAS associated with HS loading characterized a group of patients, 10% of the entire study population constituted mostly by females, with increased arterial stiffness. Data from Ricchiuti et al. 24 demonstrated that an HS diet plus RAAS activation were necessary for the development of cardiovascular damage in healthy rodents, while those animals on an LS diet showed no cardiovascular impairment in spite of a greater increase in their PRA and aldosterone levels. Hollenberg showed that rapid vascular damage requires both high mineralocorticoid levels and HS intake in high-BP diabetic patients. 25
We thought that a condition of abnormal response of the RAAS to salt load (SS) may be involved. In 1985, Redgrave et al., 26 and later in 1992, Gordon et al., 27 described a subgroup of hypertensive patients defined as nonmodulators, in whom the expected modifications in renal blood flow, BP, and natriuresis associated with a high dietary sodium intake were not observed. Most nonmodulating hypertensives do not present a normal Aldo response to administered angiotensin II, and this aberrant behavior appears to revert when angiotensin-converting enzyme inhibitors are administered.28–30 However, the exact relation between the nonmodulating condition, salt sensitivity, and RAAS has not been clearly established. A possible contribution of our analysis would be the characterization of the group represented by those hypertensive patients unable to elicit a normal RAAS response on an HS diet. As these hypertensive patients had normal creatinine and renal function, the expected response to an HS load should have been a reduction in RAAS activity. Although HS intake has been associated with a transient inhibition of RAAS in normotensives, an inadequate suppression of aldosterone in response to increased sodium intake has been described in essential hypertensive patients. Conversely, some studies suggest that in normotensive subjects and in hypertensive rats a high aldosterone secretion is a physiological response to an LS diet. 31 In this context, in the broad spectrum of responses there seems to be a complex network between salt sensitivity, RAAS, BP, kidney function, and additional mechanisms like sympathetic/parasympathetic balance, oxidative stress, and inflammatory processes.
Regarding arterial stiffness, our findings are also consistent with previous reports that associated HS intake with loss of normal vessel distensibility. In 1986, Avolio 32 demonstrated that the level of sodium intake might influence PWV independently of age, BP, and cholesterol. An enhancement of AT1 receptor expression and sensibility associated with chronic HS intake is classically considered one of the major mechanisms promoting vascular damage on HS diets. 33 Moreover, combined activation by Ang II and aldosterone of cardiac AT1 receptors, as well as the mineralocorticoid receptors, could be responsible for myocardial and vascular hypertrophy and fibrosis. Many actions of aldosterone itself are also related to vascular fibrosis and damage. Le Corvoisier et al. described a significant increase in circulating and cardiac aldosterone during HS load. 34
Safar et al. 35 in 1992 suggested that sodium might affect arterial stiffness independently from BP and proposed several arguments to explain this association involving the effect of sodium both on arterial smooth muscle tone and vasomotion.
Finally, in our study the “HS” group excreted about 120 mm/mol of Na+ daily; this would be a normal to low level in the United States (US) and also in our country, while usual salt intake is 10 to 12 gr/daily. Our study design prespecified subject salt intake classification based on the WHO recommendation. The most current WHO recommendation developed by a 2002 joint WHO/Food and Agriculture Organization of the United Nations (FAO) Expert Consultation for the general population is to reduce the consumption of sodium to below 90 mmol (< 5 g salt) per day, which must be lower for hypertensives. 23
More recent data from a Cochrane Review on longer-term effects of salt reduction published in 2004 concluded that further benefits in BP both in hypertensive and nonhypertensive individuals can occur with a further reduction of sodium intake to as low as 50 mmol (3 g salt) per day, thus raising the question of the potential need to adjust the recommended population level. 36 Based on this, we consider that although our classification of low/high sodium could not represent average sodium population consumption, it is more consistent with those levels whereas physiological responses should be preserved.
As we did not suggest changes in patients’ usual diets prior to the study, to obtain real-life sodium intake conditions and under these circumstances we found that only 52/300 (17%) of the entire population were at a recommended sodium intake rank, while 83% were on higher sodium intake with no adherence to usual recommendations. However, when we compare sodium intake from this treated population vs the general population from our country, they showed at least reduced values. Our data represent a hypertensive population and is not extrapolated to the general population, whereas the RAAS-salt relation could show a different behavior.
To summarize, although both findings described here—i.e. the inverse salt-RAAS relationship and the association between a positive salt-RAAS relation and vascular damage—were previously reported in preclinical studies, our study design allowed for their demonstration in a clinical daily practice setting.
Further research is needed to evaluate the impact of sodium intake on target organ damage and RAAS response in longer-term studies. In the meantime, an individualized evaluation of the specific sodium intake-RAAS response profile would contribute to a more rational management of salt restriction in hypertensive subjects.
Footnotes
Acknowledgements
We are grateful to Kathleen Dowd, from the Academic Development Department of the Hospital Universitario Austral, for her collaboration in the edition and translation of this manuscript.
Conflict of interest
The author declares that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.