
Abstract
Introduction:
Stretch of the atrial membrane upregulates the slow component of delayed rectifier K+ current (IKs). Blockade of angiotensin II subtype 1 receptors (AT1R) attenuates this increase in IKs. The present study aimed to examine the effects of irbesartan, a selective AT1R blocker (ABR), on both the enhancement of IKs and the shortening of action potential duration (APD) induced by stretching atrial myocytes for exploring the mechanisms underlying the prevention of atrial fibrillation (AF) by ABR.
Methods:
Hyposmotic solution (Hypo-S) was used to stretch guinea pig atrial myocytes. IKs and APD were recorded using the whole-cell patch-clamp technique.
Results:
Irbesartan (1–50 μM) attenuated the Hypo-S-induced increase in IKs and shortening of APD90. Hypo-S increased the IKs by 113.4%, whereas Hypo-S + 1 μM irbesartan and Hypo-S + 50 μM irbesartan increased the IKs by only 74.5% and 70.3%, respectively. In addition, Hypo-S shortened the APD90 by 19.0%, whereas Hypo-S + 1 μM irbesartan and Hypo-S + 50 μM irbesartan shortened the APD90 by 12.1% and 12.0%, respectively.
Conclusion:
The actions of irbesartan on electrical changes induced by stretching atrial myocytes are associated with blocking AT1R. These actions may be beneficial for treating AF.
Introduction
Increasing evidence suggests that the renin–angiotensin system (RAS) is associated with the occurrence of atrial arrhythmias in experimental animals.1–5 Recent clinical studies6–11 have also suggested that blockade of the RAS with angiotensin-converting enzyme (ACE) inhibitors or angiotensin II (Ang II) type 1 receptor (AT1R) blockers is effective for the treatment of atrial fibrillation (AF). However, mechanisms underlying the treatment are not fully understood, especially concerning actions of the drugs on electrical changes in AF.
The shortening of action potential duration (APD) and effective refractory period (ERP) are generally regarded as pivotal factors for the occurrence of reentry-based AF. During AF, impaired atrial contraction causes the atria to dilate or stretch12,13 and induce the secretion of Ang II from cardiomyocytes.14,15 Zankov et al. demonstrated that both exogenous Ang II and hyposmotic-induced membrane stretch potentiates the slow component of delayed rectifier K+ current (I Ks) in guinea pig myocytes by activating AT1R, which results in a shortened atrial APD. These results suggest that the shortening of the atrial APD, associated with IKs enhancement through the activation of AT1R, plays an important role in facilitating the initiation/maintenance of AF.16,17 Irbesartan, a selective AT1R blocker, was reported to inhibit heterologously expressed KCNQ1/KCNE1 (encoding IKs) channels, 18 which may contribute to its anti-AF mechanism. However, the effective concentrations of the drug for blocking KCNQ1/KCNE1 channels are far greater than the clinical therapeutic levels achieved in blood.19,20
In the present study, we examined the effects of irbesartan on both the increase in IKs and the shortening of APD induced by hyposmotic solution (Hypo-S) in guinea pig atrial myocytes. The results show that the actions of the drug at therapeutically relevant concentrations on electrical changes induced by the stretching of the atrial cell membrane are, at least partially, associated with blocking AT1R and therefore beneficial for AF prevention.
Materials and methods
Isolation of guinea pig atrial myocytes
The experimental procedures were conducted in accordance with the guidelines established by the Animal Care and Use Committee of Shiga University of Medical Science (Shiga, Japan). Single atrial myocytes were enzymatically dissociated from the hearts of non-pregnant adult female Hartley guinea pigs (weighing 250–350 g) using a retrograde Langendorff perfusion method as previously described. 16
Solutions and chemicals
Normal Tyrode solution (140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 0.33 mM NaH2PO4, 5.5 mM glucose, and 5.0 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH adjusted to 7.4 with NaOH) was used as “isosmotic” extracellular solution (Iso-S, average osmolality: ~285 mOsm/kg). “Hyposmotic” extracellular solution (Hypo-S, average osmolality: ~212 mOsm/kg) was prepared by simply reducing the NaCl concentration to 100 mM in the normal Tyrode solution as previously described. 17 The pipette solution contained 70 mM potassium aspartate, 50 mM KCl, 10 mM KH2PO4, 1 mM MgSO4, 3 mM Na2-ATP (Sigma), 0.1 mM Li2-GTP (Roche Diagnostics GmbH, Mannheim, Germany), 5 mM ethylene glycol tetraacetic acid (EGTA), and 5 mM HEPES, with the pH adjusted to 7.2 with KOH. Irbesartan (Dainippon Sumitomo Pharma Co., Ltd, Osaka, Japan) was dissolved in dimethyl sulfoxide (DMSO, Sigma) to yield 50 mM stock solution and diluted with Iso-S or Hypo-S to concentrations of 1 and 50 μM, respectively. The concentration of DMSO in the final solution (< 0.1%, V/V) slightly increased (< 3.8%) the osmolality of 50 μM irbesartan + Hypo-S (or Iso-S), but had no effect on cell swelling (see Supplementary Materials) or IKs.
Electrophysiological recordings and data analysis
Single atrial myocytes were either current- or voltage-clamped using the standard whole-cell patch-clamp technique with an EPC-8 patch-clamp amplifier (HEKA Electronics, Lambrecht, Germany). Data were low-pass filtered at 5 kHz, acquired at 2 kHz through a LIH-1600 analogue-to-digital converter (HEKA), and stored on a hard drive using PATCHMASTER software (HEKA). Borosilicate glass electrodes had a tip resistance of 2.5–4.0 MΩ when filled with the pipette solution. All experiments were performed at 36 ± 1°C. IKs was elicited by depolarizing voltage-clamp steps given from a holding potential of −50 mV to various test potentials, under conditions that the Na+ current was inactivated by setting the holding potential to −50 mV. The L-type Ca2+ channel current (ICa,L) and the rapid component of delayed rectifier K+ current (IKr) were blocked by 0.4 µmol/l nisoldipine (Bayer AG, Wuppertal-Elberfeld, Germany) and 0.5 µmol/l dofetilide (Sigma Chemical Co., MO, USA) added to the extracellular solution, respectively.
Variations of IKs amplitude and the time course of the IKs were determined by measuring the amplitude of tail currents elicited on repolarization to a holding potential of −50 mV following two seconds (s) of depolarization to +30 mV every 10 s. Voltage-dependence of IKs activation was evaluated by fitting the I-V relation of the tail currents to a Boltzmann equation as follows: IK,tail = 1/(1+exp((Vh–Vm)/k)), where IK,tail is the tail current amplitude, Vh is the voltage at half-maximal activation, Vm is the test potential, and k is the slope factor. The deactivation kinetics of IKs was determined by fitting a single exponential function to the tail current trace. Cell membrane capacitance (Cm) was calculated on the basis of the capacitive transients during 20 ms voltage-clamp steps (± 5 mV), using the equation Cm = τCI0/ΔVm(1–Iss/I0), where τC is the time constant of the capacitive transient, I0 is the initial peak current amplitude, Iss is the steady-state current value, and ΔVm is the amplitude of the voltage step (5 mV).
Action potentials were evoked in current-clamp mode at a rate of 0.2 Hz by suprathreshold current pulses of 2 ms duration applied through the patch electrode. The APD was measured at 90% repolarization (APD90).
All of the averaged data are presented as mean ± S.E.M. with the number of experiments shown in parentheses. Statistical comparisons were evaluated using Student’s t test or one-way analysis of variance (ANOVA) with Newman-Keuls post-hoc test, as appropriate. A p < 0.05 was considered statistically significant.
Results
Irbesartan does not affect the baseline IKs but attenuates the Hypo-S-induced IKs enhancement
Based on the concentration-dependent effect of irbesartan on KCNQ1/KCNE1 channels that was previously reported in a Chinese Hamster Ovary (CHO) expression system, 18 we chose to examine the effects of 1–50 μM irbesartan on IKs channels in guinea pig atrial myocytes, since 1–50 μM irbesartan are close to the therapeutic concentrations in blood19,20 and almost the subthreshold for the inhibition of KCNQ1/KCNE1 channels. Figure 1(a) depicts a representative time-course of atrial tail IKs during the first and second exposures to Hypo-S that caused mechanical stretch of the cell membrane and induced enhancement of the IKs. Before the second exposure to Hypo-S, the myocyte was pretreated with irbesartan for five minutes. We found that 1–50 μM irbesartan did not affect the baseline IKs of guinea pig atrial myocytes (current traces indicated by 1 and 3 in Figure 1(b)), which was quite similar to that in the CHO expression system. 18 As expected, Hypo-S induced increases in both steady-state and tail IKs currents (current traces 2 and 4 in Figure 1(b)).
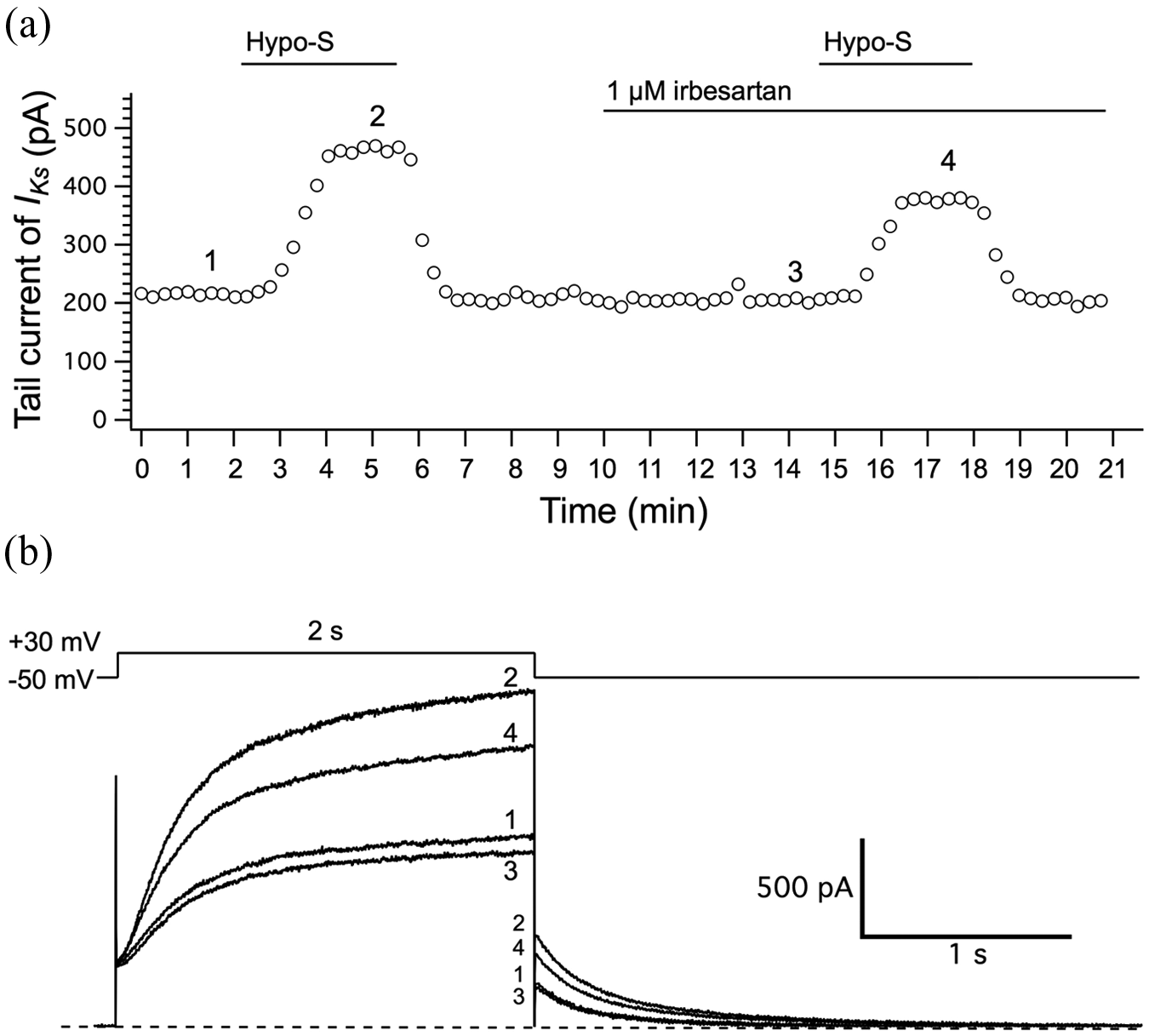
Irbesartan does not affect the baseline IKs in guinea pig atrial myocytes.
The myocyte swelling observed in Hypo-S reflects the effect of cell membrane stretching that usually occurs during the early stages of AF12,21 and affects various ion transport mechanisms, including the IKs enhancement in atrial myocytes.17,22,23 Figures 2(a) and 2(b) show typical current traces elicited by depolarizing voltage-clamp steps given from a −50 mV holding potential to various test potentials in the absence (panel (a)) or presence of 1 μM irbesartan (panel (b)) while the cells were exposed to Hypo-S. Figure 2(c) summarizes the percentage increases of tail IKs of cells in Hypo-S in the absence or presence of 1 and 50 μM irbesartan. The percentage increases in tail IKs for the control cells and those in the presence of 1 and 50 μM irbesartan were 113.40 ± 9.96% (n = 18), 74.52 ± 8.49% (n = 10), and 70.25 ± 9.34% (n = 16), respectively. Increases in tail IKs in the presence of irbesartan were significantly lower (p < 0.05) than those in the control (Figure 2(c)). Figure 2(d) shows the current-voltage relationships for tail IKs recorded during the superfusion with Iso-S (filled circles), Hypo-S (open circles), or Hypo-S + 1 μM irbesartan (open squares). The voltages for half-activation of tail IKs (Vh) were obtained by fitting the data to the Boltzmann equation and were 9.06 ± 1.28 mV (n = 22) in Iso-S, 2.08 ± 1.29 mV in Hypo-S (n = 13; p < 0.01 vs. Iso-S), and 2.10 ± 1.70 mV in Hypo-S + 1 μM irbesartan (n = 10; p < 0.05 vs. Iso-S), respectively. The Hypo-S caused a significant negative shift of Vh; however, irbesartan did not recover this negative shift in the activation gate. In addition, there were no significant differences in the parameters governing gating kinetics, irrespective of irbesartan treatment.
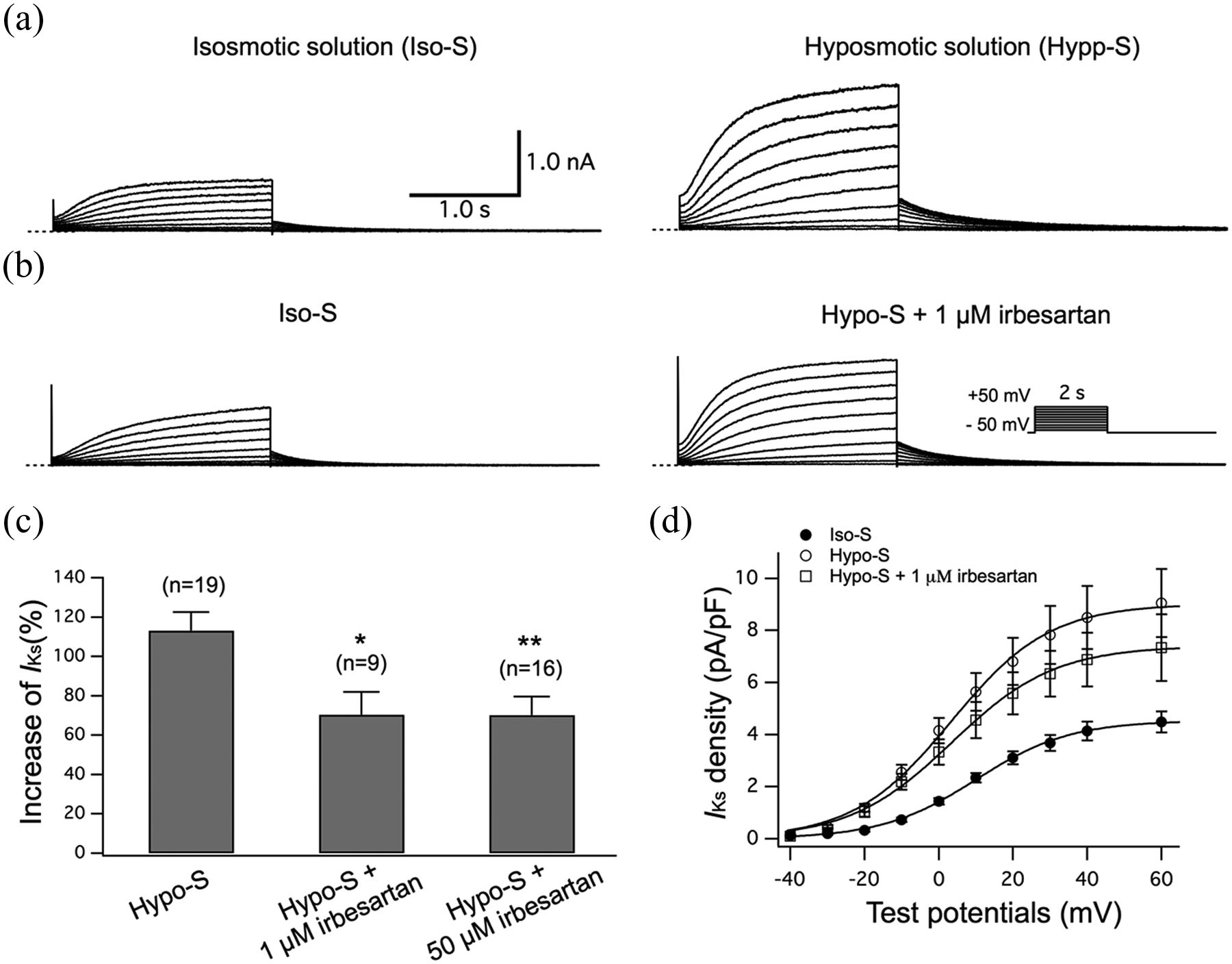
Irbesartan attenuates the Hypo-S-induced increase in IKs in guinea pig atrial myocytes. The atrial myocytes were initially superfused with control Iso-S followed by Hypo-S in the absence (a) or presence of 1 μM irbesartan (b). IKs was activated by depolarizing voltage-clamp steps given from a holding potential of −50 mV to potentials listed in the panel (b) inset. The dashed line indicates the zero current level. (c) The percentage increase in tail IKs amplitudes induced by Hypo-S without and with irbesartan (1 μM and 50 μM) at +30 mV. (d) The I-V relations for tail IKs amplitudes (expressed as current density) recorded during exposure to Iso-S (filled circles), Hypo-S (open circles), or Hypo-S + 1 μM irbesartan (open squares). Smooth curves through the data points denote the least-squares fit with the Boltzmann equation, yielding Vh (see text). *p < 0.05 and **p < 0.01 vs. Hypo-S. IKs: delayed rectifier K+ current.
Table 1 summarizes the effects of 1 μM irbesartan on the deactivation time course of tail IKs at four different test potentials. The Hypo-S significantly (p < 0.05) slowed the deactivation time course of tail IKs at voltages between −60 mV and −30 mV. There were, however, no significant differences in the increase of τ values (in parentheses) irrespective of irbesartan treatment, though there was a trend in the irbesartan-induced reduction of the Hypo-S effects on IKs deactivation.
Effect of 1 μM irbesartan on deactivation time constants (τ) of IKs in isosmotic solution (Iso-S) and hyposmotic solution (Hypo-S) at four different test potentials.

IKs: delayed rectifier K+ current; N: number of cells; Δ%, percentage increase of τ values over Iso-S; data are expressed as the mean ± S.E.M.; ap < 0.05 and bp < 0.01 vs. Iso-S.
Irbesartan attenuates Hypo-S-induced shortening of the action potential
Figure 3(a–c) shows the superimposed traces of guinea pig atrial action potentials in Iso-S and Hypo-S- (control, Figure 3(a)), Iso-S and Hypo-S + 1μM irbesartan- (Figure 3(b)), and Iso-S and Hypo-S + 50 μM (Figure 3(c)) irbesartan-treated myocytes, respectively. As the bar graphs summarize in Figure 3(d), Hypo-S shortened the APD90 by 19.03 ± 1.36 (n = 17), whereas Hypo-S + 1 μM irbesartan shortened the APD90 by only 12.05 ± 1.38 (n = 9; p < 0.01 vs. Hypo-S) and Hypo-S + 50 μM irbesartan shortened the APD90 by 12.00 ± 1.46 (n = 14; p < 0.01 vs. Hypo-S). Together, these results suggest that 1–50 μM irbesartan significantly attenuated the Hypo-S-induced shortening of action potentials in atrial myocytes. In addition, no difference in depolarized resting membrane potentials caused by Hypo-S was observed between control cells and those in the presence of irbesartan (data not shown).

Irbesartan attenuates the Hypo-S-induced shortening of APD90 in guinea pig atrial myocytes. Superimposed traces of action potentials in Iso-S followed by Hypo-S (a), Hypo-S + 1 μM irbesartan (b), or Hypo-S + 50 μM irbesartan (c), respectively. (d) The percentage decrease in APD90 after exposure to Hypo-S without and then with irbesartan (1 μM and 50 μM). Although the effects of irbesartan on the shortening of APD90 were significant, no statistical difference was observed in the resting membrane potentials. **p < 0.01 vs. control.
Discussion
Cellular electrophysiological studies have indicated that the most important impact of AF on ion channels is the marked reduction in inward I Ca,L currents, 12 which leads to atrial contractile dysfunction and induces increased membrane stretching of outward IKs currents.17,21,22,24 Changes in both ICa,L and IKs contribute to the atrial APD shortening and the loss of its physiological rate adaptation, which promotes atrial electrical and structural remodeling and creates a substrate for persistent AF.12,23
In the present study, we found that irbesartan attenuated the stretch-induced enhancement of IKs, suggesting that the drug possesses the ability to improve the pathophysiological conditions precipitating AF. This hypothesis is supported by the recent identification of KCNQ1 (encoding the α-subunit of the IKs channel) S140G and R14C mutations in familial AF cases, in which both “gain-of-function” mutations cause an enhancement of IKs.25,26 In addition, atrial membrane stretching results in an increase of IKs-mediated shortening of the atrial APD, which may facilitate the maintenance of AF.17,27,28 Irbesartan can rescue shortening of the APD that is induced by cell membrane stretching. Thus, the inhibition of extreme APD shortening as a result of atrial membrane stretching is involved in the mechanism underlying irbesartan-mediated AF prevention.
Evidence suggests that the RAS plays a pivotal role in the occurrence and maintenance of AF. Atrial membrane stretching during AF not only activates AT1R,29,30 but also induces the secretion of Ang II from cardiomyocytes.14,15 Madrid and colleagues 31 reported that irbesartan in combination with amiodarone was more effective at preventing the recurrence of AF than amiodarone alone. Several recent clinical reports6–10 and animal experiments2,32 have also confirmed the effect of AT1R blockers on AF. In the present study, therapeutically relevant concentrations of irbesartan attenuated the stretch-induced increase, but not baseline, levels of atrial IKs, suggesting that the action of the drug on electrical changes is associated with blocking AT1R. This result is consistent with a previous study 33 that found that irbesartan prevented the arrhythmogenic effect of Ang II by blocking AT1R in human atrial myocardium. Zankov et al. reported that the selective AT1R blockers valsartan and candesartan attenuate Ang II- and stretch-induced enhancement of IKs and shortening of APD, respectively, by activating AT1R in guinea pig atrial myocytes.16,17 Based on these previous findings together with the observations of irbesartan in this study, we conclude that the actions of ARBs on electrical changes associated with the dilation or stretch of the atria are involved in the AT1R blockade and are beneficial for the prevention of acute electrical remodeling during AF.
In the present study, we also found that Hypo-S depolarized resting membrane potentials in guinea pig myocytes. Since the inward rectifier current IK1 is responsible for maintaining the resting membrane potential, 23 and irbesartan did not affect the depolarization of the resting membrane potential caused by atrial membrane stretching, we postulate that IK1 is not the therapeutic target of AT1R blockers during AF. 34 This lack of effect on the resting membrane potential was also observed with candesartan in a previous study. 17
Conclusions
Irbesartan-mediated AT1R blockade attenuates the electrical changes induced by stretching atrial myocytes. This is likely why AF patients derive a pronounced benefit with ABRs such as irbesartan.
Footnotes
Acknowledgements
We thank Dainipon Sumitomo Pharma Co., Ltd (Osaka, Japan) and Sanofi-Aventis (France) for kindly providing us with irbesartan as a reagent.
Conflict of interest
None declared.
Funding
This work was supported, in part, by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (to MH); National Natural Science Foundation of China (#81273501 to JW and WGD, and #30930105 to WJZ); Major International (Regional) Joint Research Project of National Natural Science Foundation of China (#81120108002 to WJZ); Uehara Memorial Foundation; and the Ministry of Health, Labor and Welfare of Japan for Clinical Research on Measures for Intractable Diseases (to MH).