
Abstract
This study aimed to examine the protective effects of aliskiren on gentamicin-induced nephropathy. Rats were injected with gentamicin (100 mg/kg per day) for 14 days. Aliskiren was infused for two weeks. Human proximal tubular epithelial cell lines (HK-2) were cultured with gentamicin in the absence or presence of aliskiren. Inflammatory profibrotic and apoptotic markers were evaluated in vivo and in vitro. Aliskiren treatment attenuated the decreased creatinine clearance, increased fractional sodium excretion, glomerulosclerosis and tubulointerstitial fibrosis and counteracted the increased ED-1 expression in gentamicin-treated rats. The levels of inflammatory cytokines (TNF-α, IL-1β and IFN-γ) and adhesion molecules (MCP-1, ICAM-1 and VCAM-1) increased in the gentamicin-treated kidneys. These changes were restored by aliskiren co-treatment. Aliskiren effectively reversed transforming growth factor-β-induced fibrotic responses such as induction of α-smooth muscle actin in gentamicin-treated rat kidneys. Along with these changes, aliskiren also attenuated the increase in nuclear factor κB and phosphorylated extracellular signal-regulated kinase (pERK 1/2) levels in HK-2 cells cultured with gentamicin. In addition, aliskiren decreased the number of TUNEL-positive nuclei and reduced the expression of proapoptotic markers in gentamicin-treated HK-2 cells. These findings suggest that aliskiren attenuates gentamicin-induced nephropathy by suppression of inflammatory, profibrotic and apoptotic factors through inhibition of the nuclear factor κB, Smads and mitogen-activated protein kinase signaling pathways.
Introduction
In progressive renal diseases, including gentamicin (GM)-induced nephropathy, renal inflammation is a vital pathologic process in the evolution of chronic kidney disease; this inflammation is characterized by the infiltration of inflammatory cells such as monocytes/macrophages and myofibroblasts.1,2 Infiltrating macrophages in the renal tubular interstitium can release proinflammatory chemoattractive cytokines, thereby leading to the formation of a vicious self-accumulation cycle. Furthermore, infiltrating macrophages can produce profibrotic cytokines such as transforming growth factor (TGF)-β1, which plays a crucial role in progressive renal fibrosis.3,4
Multiple signaling pathways are thought to mediate these inflammatory and fibrotic processes in progressive renal diseases. One of these pathways, the nuclear factor κB (NF-κB) pathway, is known to regulate the expression of numerous genes involved in inflammation and cell proliferation. Thus, it is thought to be a key transcription factor that can mediate acute and chronic inflammation by regulating the gene expression of cytokines, chemokines and adhesion molecules and consequently cause apoptosis and interstitial fibrosis in progressive renal disease.5,6
Additionally, in vitro studies have suggested that activation of the extracellular signal-regulated kinase (ERK) pathway also plays a critical role in the proliferation of tubular epithelial and myofibroblast-like cells. 7 Mitogen-activated protein kinases (MAPKs) use three parallel signal transduction pathways. Cell proliferation is triggered via transient ERK activation and differentiation is triggered via sustained ERK activation; however, the exact role of the ERK signaling pathway in progressive renal disease remains to be determined. GM also directly activates apoptotic genes and increases apoptosis in tubular and interstitial cells, resulting in tubular atrophy. 8
The autocrine and paracrine activities of the tissue renin–angiotensin system (RAS) are important modulators of the structure and function of a number of organs and systems, including the heart, kidneys, vasculature and skeletal muscle. 9 Aliskiren, a low molecular weight, nonpeptide, direct renin inhibitor (DRI), which has demonstrated targeted organ protection in various animal models,10,11 is now available for treating human hypertension. 12 However, it is not known whether aliskiren treatment in GM-induced nephropathy leads to organ protection.
In this study, we investigated the effects of aliskiren on GM-induced nephropathy; to elucidate its mechanism of action, we examined the effect of aliskiren on renal inflammation, fibrosis and apoptosis after GM-induced renal injury.
Materials and methods
Animals
Male Sprague–Dawley rats weighing 180–200 g were used. The experimental procedure conformed to the Institutional Guidelines for Experimental Animal Care and Use. The rats were treated with GM (100 mg/kg per day, intramuscularly (IM)) alone, GM with aliskiren (Novartis, USA, 25 mg/kg per day) using osmotic minipumps (models 2002, Alzet, Palo Alto, CA, USA) or vehicle for 14 days. The rats were maintained individually in the metabolic cages for the last three days to allow urine collections for the measurement of Na+ and creatinine. On the day of the experiment, under anesthesia with isoflurane, blood samples were collected from the inferior vena cava and analyzed for Na+ and creatinine. The right kidney was rapidly removed and processed for immunoblotting as described below. The left kidney was fixed via retrograde perfusion for immunohistochemistry.
In another set of experiments, the rats were decapitated under a conscious state and the kidneys were removed and kept at −70°C until assayed for mRNA expression by reverse transcriptase-polymerase chain reaction (RT-PCR) and real-time polymerase chain reaction (real-time PCR).
Measurement of GM concentration in whole kidney
GM was extracted from renal tissues by using a sodium hydroxide (NaOH) extraction method. Briefly, 300 μl of phosphate-buffered saline (PBS) was added to the tube, and the contents were homogenized. The homogenizer was rinsed with another 300 μl of PBS, which was pooled with the original homogenate. An equal volume of 2M NaOH was added to the homogenate, and the mixture was incubated at 70°C for 20 min. Samples were centrifuged at 13,000 g for 20 min, and the supernatant was collected and neutralized to pH 7.0 before GM concentration was measured. GM concentrations were determined using a fluorescence polarization immunoassay.13,14
Cell culture and application of GM and aliskiren to HK-2 cells
An immortalized proximal tubule epithelial cell line from HK-2 was purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured. Briefly, cells were passaged every 3–4 days in 100-mm dishes (Falcon, Bedford, MA, USA) using Dulbecco’s modified Eagle’s medium-F12 (Sigma Chemical Co., St Louis, MO, USA) supplemented with 10% fetal bovine serum (Life Technologies Inc., Gaithersburg, MD, USA), insulin-transferrin-sodium selenite media supplement (Sigma Chemical Co., St Louis, MO, USA), 100 U/ml penicillin and 100 mg/ml streptomycin (Sigma Chemical Co., St Louis, MO, USA). These cells were incubated in a humidified atmosphere of 5% CO2, 95% air at 37ºC for 24 h and subcultured at 70–80% confluence. For experimental use, HK-2 cells were plated onto 60-mm dishes in medium containing 10% fetal bovine serum for 24 h and cells were then switched to Dulbecco’s modified Eagle’s medium-F12 with 2% fetal bovine serum for 16 h. These cells were treated with GM (0.5 and 1.0 mg/ml) in the presence or absence of aliskiren (50 and 100 nmol). Control cells received buffer only instead of GM or aliskiren. The cells were harvested at the end of the treatment for further analysis.
Semiquantitative immunoblotting
The dissected kidney was homogenized in ice-cold isolation solution containing 0.3 M sucrose, 25 mM imidazole, 1 mM EDTA, 8.5 µM leupeptin, 1 mM phenylmethylsulfonyl fluoride, pH 7.2. The homogenates were centrifuged at 1000 g for 15 min at 4°C to remove whole cells, nuclei and mitochondria. The total protein concentration was measured (Pierce BCA protein assay reagent kit, Pierce, Rockford, IL, USA). All samples were adjusted with isolation solution to reach the same final protein concentrations and solubilized at 65°C for 15 min in SDS-containing sample buffer and then stored at −20°C. To confirm equal loading of protein, an initial gel was stained with Coomassie blue. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed on 9 or 12% polyacrylamide gels. The proteins were transferred by gel electrophoresis (Bio-Rad Mini Protean II, Bio-Rad, Hercules, CA, USA) onto nitrocellulose membranes (Hybond ECL RPN3032D, Amersham Pharmacia Biotech, Little Chalfont, UK). The blots were blocked with 5% milk in PBS-T (80 mM Na2HPO4, 20 mM NaH2PO4, 100 mM NaCl and 0.1% Tween 20, pH 7.5) for 1 h and then incubated overnight at 4°C with primary antibodies, followed by incubation with secondary anti-rabbit, anti-mouse or anti-goat horseradish peroxidase-conjugated antibodies. The labeling was visualized using an enhanced chemiluminescence system.
Immunohistochemistry
A perfusion needle was inserted in the abdominal aorta and the vena cava was cut to establish an outlet. Blood was flushed from the kidney with cold PBS (pH 7.4) for 15 s before switching to cold 3% paraformaldehyde in PBS (pH 7.4) for 3 min. The kidney was removed and sectioned into 2–3 mm transverse sections and immersion fixed for 1 h, followed by 3 × 10 min washes in PBS. The tissue was dehydrated in a graded ethanol series and left overnight in xylene. After embedding in paraffin, 2 µm sections were made on a rotary microtome. Immunoperoxidase labeling was performed as previously described. 15
Real-time polymerase chain reaction (real-time PCR)
Renal cortex was homogenized in Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA was extracted with chloroform, precipitated with isopropanol, washed with 75% ethanol and then dissolved in distilled water. The RNA concentration was determined by the absorbance read at 260 nm (Ultraspec 2000; Pharmacia Biotech, Cambridge, UK). The mRNA expression of inflammatory cytokines and adhesion molecules was determined by real-time PCR. cDNA was made by reverse transcribing 5 μg of total RNA using oligo(dT) priming and superscript reverse transcriptase II (Invitrogen, Carlsbad, CA, USA). cDNA was quantified using Smart Cycler II System (Cepheid, Sunnyvale, CA, USA) and SYBR Green was used for detection. Each PCR reaction was done in 10 μM forward primer, 10 μM reverse primer, 2X SYBR Green Premix Ex Taq (Takara Bio Inc., Japan), 0.5 μl cDNA and H2O to bring the final volume to 20 μl. Relative levels of mRNA were determined by real-time PCR, using a Rotor-GeneTM 3000 Detector System (Corbette Research, Mortlake, New South Wales, Australia). Sequences of primers are listed in Table 1.
Primer sequences for real-time polymerase chain reaction.

TNF: tumor necrotic factor; IL: interleukin; IFN: interferon; MCP: monocyte chemoattractant protein; ICAM: intercellular adhesion molecule; VCAM: vascular cell adhesion molecule; Fwd: forward; Rev: reverse.
The PCR was performed according to the following steps: 1) 95°C for 5 min; 2) 95°C for 20 s; 3) 58–62°C for 20 s (optimized for each primer pair); 4) 72°C for 30 s; and 5) 85°C for 6 s to detect SYBR Green. Steps 2–5 were repeated for additional 64 cycles, while at the end of the last cycle temperature was increased from 60°C to 95°C to produce a melt curve. Data from the reaction were collected and analyzed with the Corbett Research Software. The comparative critical threshold (Ct) values from quadruplicate measurements were used to calculate the gene expression, with normalization to GAPDH as an internal control. 16 Melting curve analysis was performed to enhance specificity of the amplification reaction.
Pathologic examinations
The extent of glomerulosclerosis (GS) was graded from 0 to 4 by a semiquantitative score: 0: normal; 1: mesangial expansion/sclerosis involving <25% of the tuft; 2: moderate GS (25 to 50%); 3: severe GS (50 to 75%); and 4: diffuse GS involving >75% of the glomerular tuft. The glomerulosclerosis index (GSI) for each rat was calculated as a mean value of all glomerular scores obtained. 17 Tubulointerstitial lesion indexes were determined using a semiquantitative scoring system. 17 Ten fields per kidney were examined, and lesions were graded from 0 to 3 (0: no change; 1: changes affecting <25% of the section; 2: changes affecting 25 to 50% of the section; and 3: changes affecting 50 to 100% of the section) according to the area with tubulointerstitial lesions (tubular atrophy, casts, interstitial inflammation, and fibrosis). The score index in each rat was expressed as a mean value of all scores obtained.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
The ApopTag in situ apoptosis detection kit (Oncor, Gaithersburg, MD, USA) was used. The sections were dewaxed and treated with proteinase K, then incubated with equilibration buffer for 10 min, followed by incubation with working-strength TdT enzyme solution at 37°C for 2 h. The reaction was terminated by incubation in working-strength stop/wash buffer for 30 min at 37°C. Sections were then incubated with antidigoxigenin peroxidase and then incubated with diaminobenzidine and 0.01% H2O2 for 5 min at room temperature. The sections were counterstained with hematoxylin and examined by light microscopy. 18
Primary antibodies
Anti-rabbit antibodies against TGF-β1 (polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-p44/42 MAPK (pERK 1/2; Thr202/Tyr204; monoclonal; Cell Signaling Technology, MA, USA), IκB-α (polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA), Bax, Bcl2, total caspase-3, cleaved caspase-3, Smad-2/3, Smad-4, Smad-6 (Cell Signaling Technology, MA, USA), phospho-NF-κB p65 (p65 NF-κB; Ser536; monoclonal; Cell Signaling Technology, MA, USA) and renin receptor (Sigma Chemical Co., St Louis, MO, USA), and anti-mouse antibodies against ED-1 (monoclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA), α-SMA (1A4 Clone; monoclonal; Sigma Chemical Co., St Louis, MO, USA) and fibronectin (2Q604; monoclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were commercially obtained.
Statistical analyses
Results are expressed as mean ± SE. Multiple comparisons among the groups were made by one-way ANOVA and post hoc Tukey HSD test. Differences with values of p<0.05 were considered significant.
Results
Functional parameters
Table 2 presents changes in functional parameters. GM treatment resulted in increased plasma creatinine levels and urine output. Fractional sodium excretion (FENa) also increased. Aliskiren treatment ameliorated increased plasma creatinine levels and also ameliorated the urine output in GM-treated rats.
Changes in renal functional parameters.
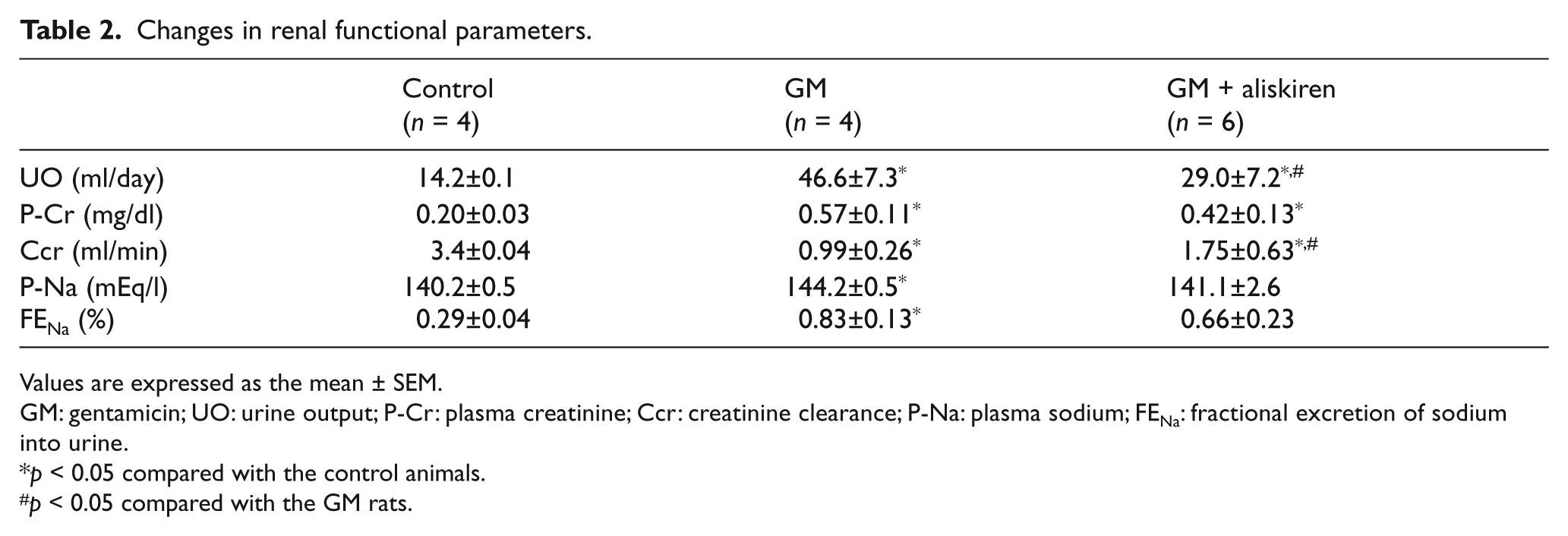
Values are expressed as the mean ± SEM.
GM: gentamicin; UO: urine output; P-Cr: plasma creatinine; Ccr: creatinine clearance; P-Na: plasma sodium; FENa: fractional excretion of sodium into urine.
p < 0.05 compared with the control animals.
p < 0.05 compared with the GM rats.
Effects of aliskiren on GM concentration and renin receptor expression
Table 3 presents changes of GM concentration in whole kidney. There was no significant difference in GM concentration between GM or aliskiren co-treatment in GM-treated rats. This finding suggests that the effect of aliskiren was not attributed to the competitive inhibition of GM accumulation in the kidney. The protein expression of the renin receptor was not changed by GM or aliskiren co-treatment in GM-treated rats compared with control rats (Figure 1).
Concentration of gentamicin in whole kidney.

Values are expressed as the mean±SEM.
GM: gentamicin.
p < 0.05 compared with the control animals.
p < 0.05 compared with the GM rats.

Semiquantitative immunoblotting of renin receptor. The protein expression of renin receptor was not changed by GM or aliskiren co-treatment in GM-treated rats compared with control rats.
Effects of aliskiren on pathological changes in GM-induced renal injury
Figure 2 shows the morphological changes observed in the kidneys among the three groups of animals. Hematoxylin and eosin staining indicated the presence of tubular casts, obstructions and vessel dilatations in the rats with GM-induced renal injury. The GSI was higher in the rats with GM-induced renal injury than in the control rats; treatment with aliskiren was associated with a less pronounced increase in GSI. In addition, interstitial expansion was also a prominent component of the renal injuries in the rats with GM-induced renal injury, which was attenuated by aliskiren treatment.
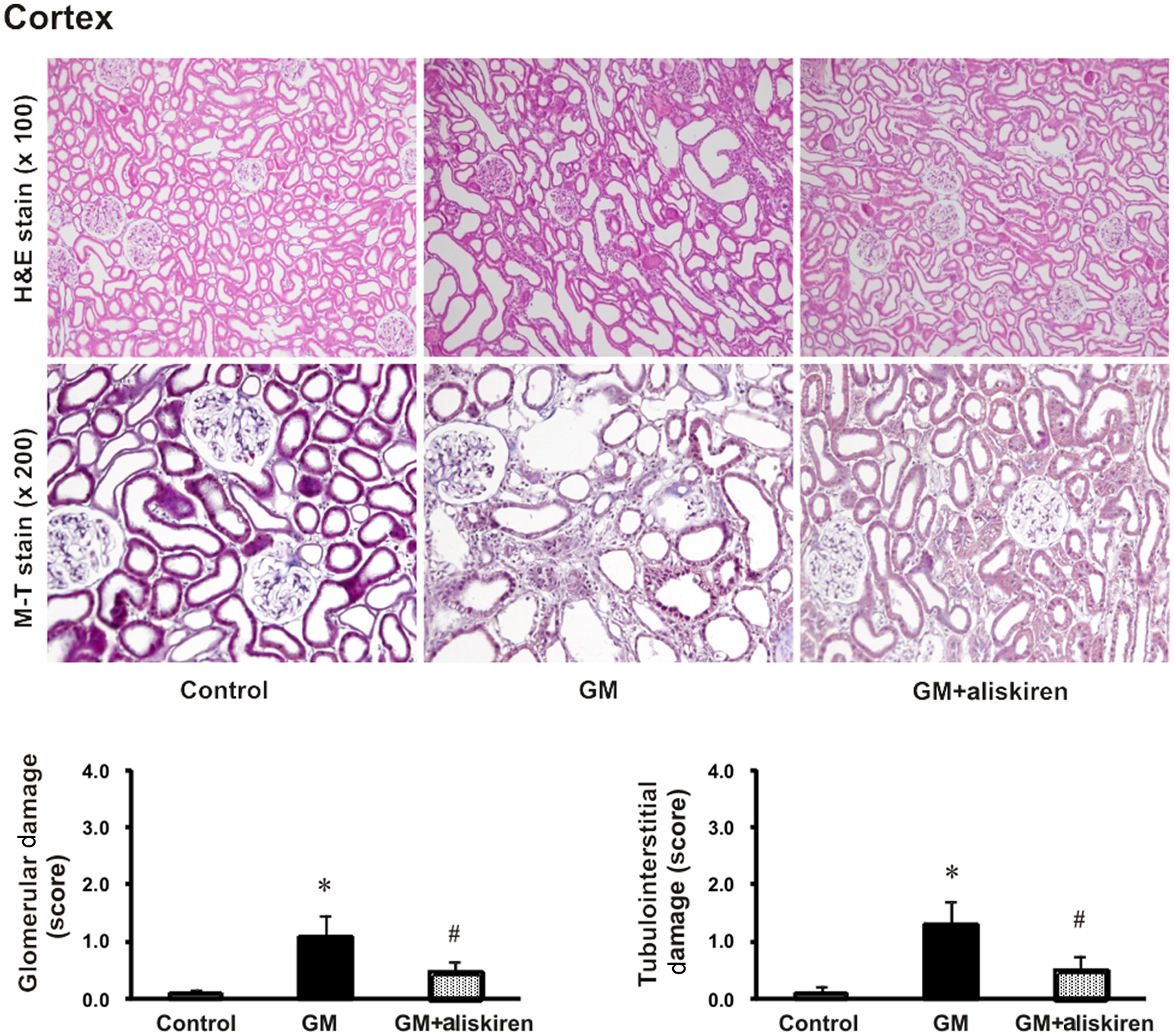
Hematoxylin and eosin (H&E) stain and Masson’s trichrome (M-T) stain of the renal cortex. The extent of glomerulosclerosis and interstitial fibrosis was higher in the rats with gentamicin (GM)-induced renal injury than in the control animals. These effects were attenuated by aliskiren treatment.
Effects of aliskiren on inflammatory cell infiltration in GM-induced renal injury
For determining the effects of aliskiren on renal inflammatory cell infiltration, the protein expression of ED-1 and iNOS and the infiltration of ED-1-positive macrophages in renal tissue were analyzed (Figure 3). The protein expression of ED-1 and iNOS in the kidney was significantly higher in GM-treated rats than in the controls and was ameliorated by aliskiren treatment (Figure 3(a)). The infiltration of ED-1-positive macrophages in GM-treated rats was significantly higher than that in the control rats. Aliskiren co-treatment abrogated inflammatory cell infiltration in GM-treated kidneys (Figure 3(b)).
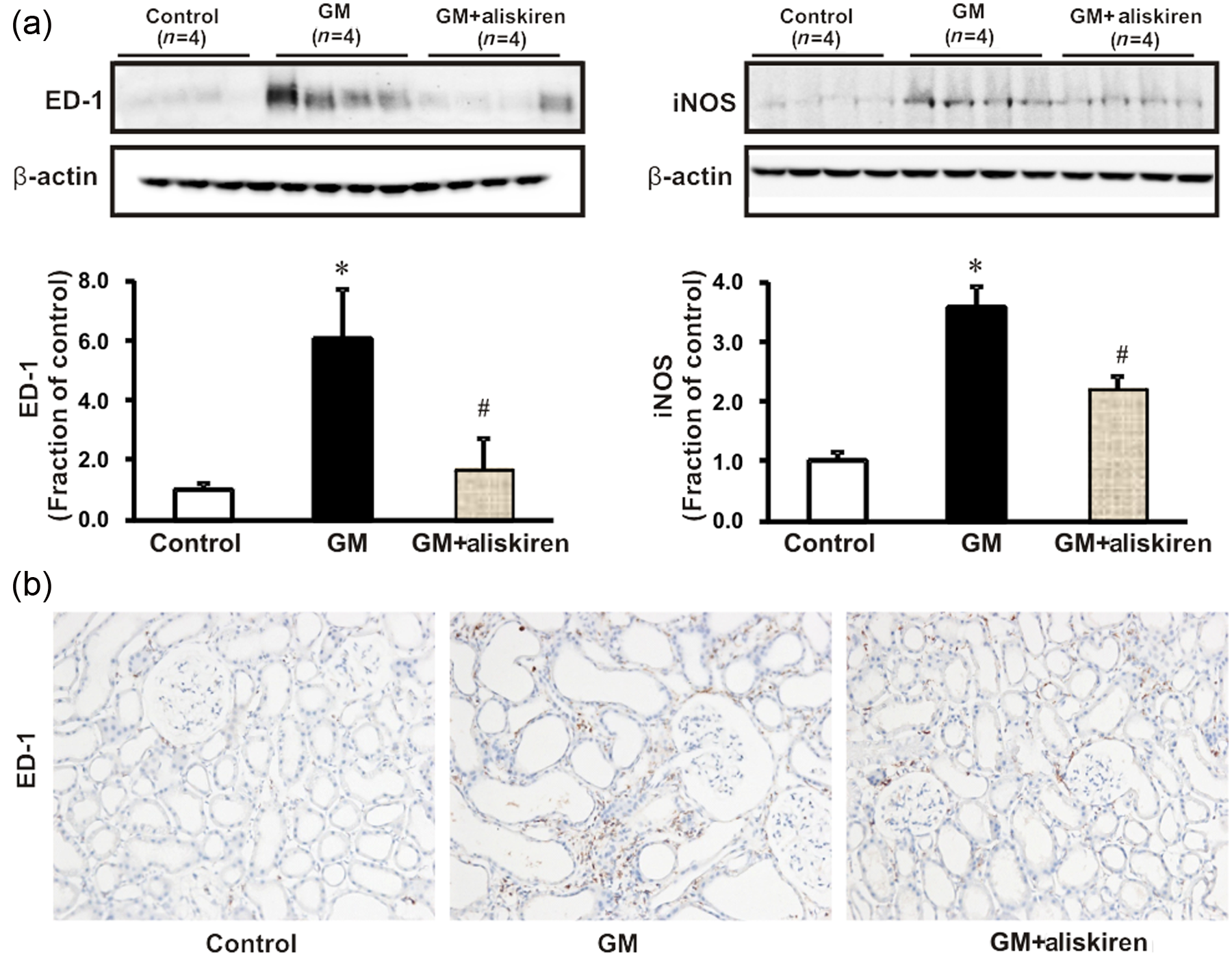
(a) Semiquantitative immunoblotting of ED-1 and inducible nitric oxide synthase (iNOS) in the kidney. Densitometric analysis showed higher ED-1 expression in the rats with gentamicin (GM)-induced renal injury than in the control rats; the increase was counteracted by aliskiren treatment. *p < 0.05 compared with the control rats; #p < 0.05 compared with the GM rats. (b) Immunoperoxidase microscopic images of ED-1 in the renal cortex. Increased immunolabeling was evident in the GM rats; the increase was prevented with aliskiren treatment. Magnification: ×200.
Effects of aliskiren on the expression of inflammatory cytokines and adhesion molecules in GM-induced renal injury
We also investigated the expression of TNF-α, IL-1β and IFN-γ, which are key inflammatory cytokines that are produced by infiltrating cells. As shown in Figure 4, GM treatment significantly induced renal TNF-α, IL-1β and IFN-γ mRNA expression, while these changes were attenuated with aliskiren co-treatment.

Real-time polymerase chain reaction of tumor necrotic factor (TNF)-α, interleukin (IL)-1β and interferon (IFN)-γ, which are proinflammatory cytokines participating in the pathogenesis of renal disease. Chemoattractants and adhesion molecules such as monocyte chemoattractant protein (MCP)-1, intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 were also evaluated. Aliskiren co-treatment suppressed the overexpression of these inflammatory cytokines and adhesion molecules induced by gentamicin (GM).
Increased expression of certain chemokines and adhesion molecules such as MCP-1, ICAM-1 and VCAM-1, which can activate, recruit or induce the transmigration of inflammatory cells into the site of renal injury, was also seen. The expression of these factors was induced by GM treatment. Aliskiren co-treatment significantly reduced the expression of these chemokines in the GM-treated rat kidneys (Figure 4).
Effects of aliskiren on NF-κB expression in GM-induced renal injury
Figure 5 shows the changes in the expression of the NF-κB p65 subunit in nuclear extracts of renal tubular HK-2 cells incubated with GM (1.0 mg/ml). Expression of the p65 NF-κB subunit increased at 1 h after GM exposure. NF-κB expression was also determined in HK-2 cells that were pretreated with aliskiren (50 and 100 nmol) for 3 h after GM (1.0 mg/ml) exposure for 6 h. Expression of the p65 NF-κB subunit was higher with GM than in the control, while the increase induced by GM was attenuated with aliskiren pretreatment.

The expression of the p65 subunit of nuclear factor κB (NF-κB) and cytosol IκBα was assessed in HK-2 cells incubated with gentamicin (GM) (1.0 mg/ml) by semiquantitative immunoblotting. The nuclear p65 subunit expression was analyzed in HK-2 cells incubated with GM (1.0 mg/ml) for 6 h in the absence and presence of aliskiren. Aliskiren (50 and 100 nmol/l) was used to pretreat HK-2 cells 3 h before GM exposure. The GM-induced overexpression of the nuclear p65 subunit of NF-κB was ameliorated with aliskiren (100 nmol/l) pretreatment.
Effects of aliskiren on pERK 1/2 expression in GM-induced renal injury
When HK-2 cells were incubated with GM (1.0 mg/ml), pERK 1/2 antibody binding increased 30 min after GM exposure and remained high over time (data not shown). pERK and pP38 expression was also determined in HK-2 cells pretreated with aliskiren (50 and 100 nmol) at 2 h before GM administration. In this experiment, pERK and pP38 overexpression induced by GM was significantly repressed by aliskiren (100 nmol) pretreatment. pJNK expression did not change on treatment with GM or aliskiren (Figure 6).

Mitogen-activated protein kinase expression was also analyzed in HK-2 cells incubated with gentamicin (GM) (1.0 mg/ml) for 3 h in the absence and presence of aliskiren. Aliskiren (50 and 100 nmol/l) was pretreated in HK-2 cells 2 h before GM exposure. Note the significantly decreased pERK 1/2 and p38 expression in the GM with aliskiren (50 and 100 nmol/l) groups compared with that for the GM group.
Effects of aliskiren on TGF-β1 and Smad expression in GM-induced renal injury
Semiquantitative immunoblotting showed that the expression of TGF-β1, an important profibrotic molecule derived from infiltrating inflammatory cells, significantly increased in GM-treated rats and was markedly attenuated by aliskiren treatment. We also assessed the TGF-β1-triggered Smad signaling pathway by evaluating the total Smad-2/3 and Smad-4 levels and the level of inhibitory Smad-6. An increase in the total Smad-2/3 level in GM-treated rats was accompanied by an increase in Smad-4 content and a decrease in the inhibitory Smad-6 content compared with those in the control group. These GM-induced changes were again restored by aliskiren co-treatment (Figure 7).
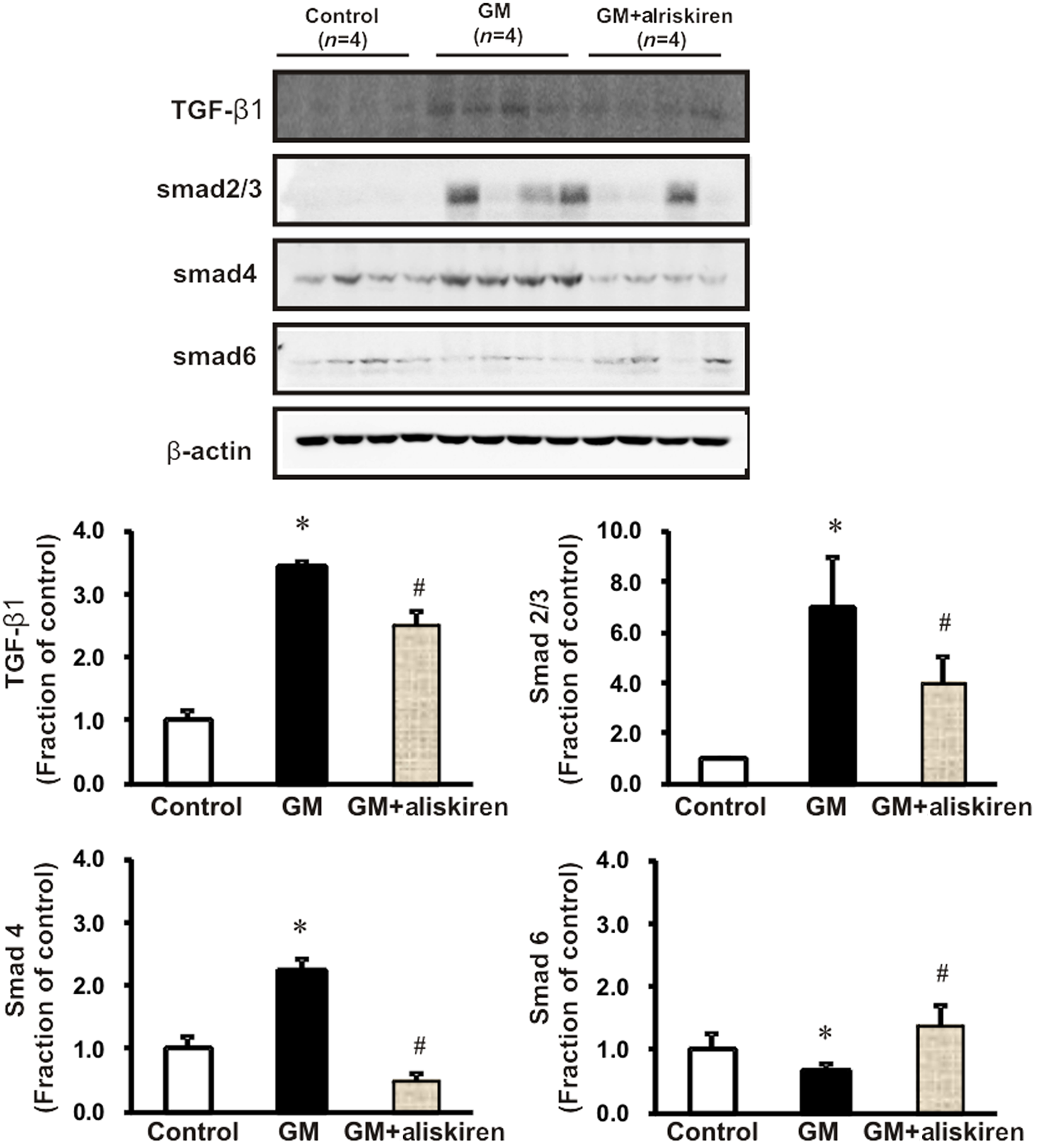
Semiquantitative immunoblotting for transforming growth factor (TGF)-ß1 and Smad-2/3, Smad-4 and Smad-6 proteins. TGF-ß1 expression increased significantly in the kidneys of gentamicin (GM)-treated rats, and was markedly attenuated by aliskiren co-treatment. Consistent with the changes in TGF-ß1, increases in Smad-2/3 and Smad-4 protein expression suggest that TGF-ß1 triggered the Smad signaling cascade. The expression of inhibitory Smad-6 was lower in GM-treated rats than in the control group. These GM-induced changes were inhibited by aliskiren co-treatment. Magnification: ×200.
Effects of aliskiren on renal fibrosis in GM-induced renal injury
We next examined the expression of fibronectin and α-smooth muscle actin (α-SMA), the molecular marker of myofibroblasts. Figure 8 shows that GM treatment significantly increased the protein expression of fibronectin and α-SMA. These changes were ameliorated by aliskiren treatment (Figure 8(a)). In vitro studies showed that aliskiren pretreatment also reduced fibronectin expression induced by GM (Figure 8(b)).

(a) Semiquantitative immunoblotting and immunohistochemistry of fibronectin and α-smooth muscle actin (SMA), a molecular marker of myofibroblasts. Protein expression of fibronectin and SMA increased significantly in the kidney of gentamicin (GM)-treated rats, and was markedly attenuated by aliskiren co-treatment. *p < 0.05 compared with the control animals; #p < 0.05 compared with the GM rats. Magnification: ×200. (b) Fibronectin expression analyzed in HK-2 cells incubated with GM (1.0 mg/ml) for 3 h in the absence and presence of aliskiren. Aliskiren (50 and 100 nmol) was pretreated in HK-2 cells 2 h before GM exposure. Note the significantly decreased fibronectin expression in the GM with aliskiren (50 and 100 nmol) groups compared with that for the GM group. *p < 0.05 compared with the control; #p < 0.05 compared with GM-treated HK-2 cells.
Effects of aliskiren on renal tubular cell apoptosis in GM-induced renal injury
GM treatment increased the expression of the proapoptotic marker Bax and decreased that of the antiapoptotic protein Bcl-2, resulting in an overall increase in the Bax/Bcl-2 ratio. Aliskiren treatment attenuated the increase in the Bax/Bcl-2 ratio in GM-treated rats (Figure 9(a)). To determine the protective effects of aliskiren on GM-induced renal tubular apoptosis, we performed TUNEL. The number of tubular epithelial cells containing TUNEL-positive nuclei increased in GM-treated rat kidneys; aliskiren co-treatment attenuated this effect (Figure 9(b)). Furthermore, the Bax and cleaved caspase-3 levels increased in GM-pretreated HK-2 cells, which was also attenuated by aliskiren treatment (Figure 10).

Expression of proapoptotic Bax and antiapoptotic Bcl-2. The Bax/Bcl-2 ratio increased in gentamicin (GM)-treated rats and was attenuated by aliskiren co-treatment. *p < 0.05 compared with the control animals; #p < 0.05 compared with the GM rats. (b) Effects of aliskiren treatment on tubular cell apoptosis in rats. The terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay showed increased apoptosis in response to GM, whereas aliskiren co-treatment significantly reduced the number of TUNEL-positive cells. Magnification: ×200.

Effects of aliskiren on the expression of pro- and antiapoptotic markers. Semiquantitative immunoblotting indicated that the level of the proapoptotic marker Bax increased in gentamicin (GM)-treated HK-2 cells. Significant increases in cleaved caspase-3 expression were noted in GM-treated cells. Aliskiren also attenuated all of these changes.
Discussion
In the present study, aliskiren treatment did not change the GM concentration and renin receptor expression in the kidney. This finding suggests that the effect of aliskiren was attributed neither to the competitive inhibition of GM accumulation in the kidney nor to the downregulation of renin receptor expression. It has been reported that there was no difference between the affinity of angiotensin II for its receptor between glomeruli from controls and those from rats treated with GM. 19 These findings do not support the role of angiotensin II in the development and maintenance of GM-induced renal injury. Therefore, the renoprotective effects of aliskiren might be pleiotropic.
A decline in renal function in chronic kidney disease often correlates with the extent of inflammation. 20 GM, in particular, can accumulate in the renal cortex, promoting the interstitial proliferation of fibroblast cells and focal infiltration by inflammatory cells. 21 In this study, GM-treated rats showed a marked increase in monocyte/macrophage infiltration into the renal cortex/medulla, as indicated by the large number of ED-1-positive cells in the interstitium. These findings are consistent with previous observations demonstrating that GM can cause an increase in monocyte/macrophage populations in the kidney. 22 GM increased the expression of proinflammatory markers such as TNF-α, IL-1β and IFN-γ. These inflammatory molecules participate in the pathogenesis of tubulointerstitial impairment via the promotion of leukocyte attraction and adhesion to inflamed renal tubular cells. The expression of cell surface adhesion molecules such as MCP-1, ICAM-1 and VCAM-1, which are highly specific chemotactic factors for macrophages, increased in the kidneys of GM-treated rats. These findings also indicate that the inflammatory process plays a significant role in the pathogenesis of GM-induced renal injury. Furthermore, we showed that aliskiren significantly reduced the infiltration of ED-1-expressing macrophages in the kidney and decreased the GM-induced renal expression of proinflammatory cytokines and cell surface adhesion molecules. These findings suggest that aliskiren can attenuate the kidney damage by suppression of cell surface adhesion molecules as well as inflammatory cytokines.
NF-κB is thought to be a key transcription factor that underlies the renal inflammatory process by regulating the gene expression of cytokines, chemokines and adhesion molecules in progressive renal diseases. In the GM rat model, NF-κB was activated by GM administration, and blockade of NF-κB activation reduced apoptosis and interstitial fibrosis. 23 NF-κB is released from an inhibitory subunit I-κB and translocates into the nucleus, where it promotes the transcriptional activation of target genes. 6 In our study, the expression of nuclear p65 subunits of NF-κB increased after GM treatment in HK-2 cells, which suggests that GM induced NF-κB activation and translocation via I-κB degradation. A recent study also suggested that aliskiren can repress NF-κB expression in diabetic rats. 24
Multiple possible intracellular mechanisms are known to regulate inflammation and fibrosis cascades in diseased kidneys. TGF-ß1 is a key molecule in these processes, and a comprehensive survey indicates that in tubular epithelial cells it is capable of activating several signal transduction pathways such as those involving MAPKs. 25 MAPKs are fundamental regulators of most immune cell functions, including proliferation, differentiation, survival and apoptosis, chemoattraction and inflammatory mediator production. They use three parallel signal transduction pathways, that is, those involving ERK, c-Jun NH2-terminal kinase and p38. One of these pathways, the ERK signaling pathway, has been shown to be activated by TGF-ß in mesangial cells. 26 TGF-ß increases ERK activity and fibronectin expression in rat mesangial cells. 27 In addition, specific inhibition of ERK prevented TGF-ß-induced fibronectin expression. These results indicate that the ERK pathway may play an important role in TGF-ß-mediated fibrosis. Other researchers have suggested that activation of the ERK pathway also plays a critical role in the proliferation of tubular epithelial and myofibroblast-like cells. 7 We also observed that ERK 1/2 activation was induced within several hours of GM administration to HK-2 cells, and this increase was attenuated by aliskiren. These changes coincide with the initiation of the fibrotic process, which has been shown to include overexpression of fibronectin and α-SMA in vivo and in vitro.
TGF-ß1 signals are transduced by transmembrane serine/threonine kinase type I and type II receptors and intracellular mediators known as Smads. On TGF-ß1 stimulation, receptor-bound Smad proteins such as Smad-2/3 are phosphorylated. Phosphorylation induces the association of Smad-2/3 with Smad-4, a member of the co-Smad subfamily, and they form transcriptionally active complexes that translocate into the nucleus and activate the transcription of TGF-ß-induced target genes. Smad signaling can also be negatively controlled by the inhibitory Smad-6 and Smad-7 proteins. Therefore, TGF-ß1-induced fibrosis is thought to be mediated by Smad signaling. 4 In our study, GM induced the overexpression of Smad-2/3 and Smad-4, whereas inhibitory Smad-6 expression decreased in response to GM. ERK and PI3K signaling may be involved in GM-induced reactive oxygen species generation and subsequent renal cell damage. 28 We showed that aliskiren reduced ERK 1/2 and p38 activation in GM-treated rat kidneys. These changes coincided with the in vivo and in vitro expression of renal fibrosis markers such as fibronectin and α-SMA. Therefore, the ERK 1/2 and p38 signaling pathways activated by TGF-ß1 appear to mediate GM-induced renal fibrosis, which is effectively inhibited by aliskiren. Masson’s trichrome staining confirmed aliskiren-induced attenuation of collagen deposition and fibrosis in GM-induced nephropathy.
In progressive renal disease, tubular cell apoptosis precedes the manifestations of tubular atrophy, tubular dilatation and perivascular inflammation. 4 GM induces Bax aggregation and translocation to the mitochondria, causing activation of caspase-9, which then cleaves and activates the effector caspase, caspase-3, leading to a loss of mitochondrial transmembrane potential and to apoptotic cell death. 29 In this study, the number of TUNEL-positive cells increased after GM treatment. Along with these changes, GM increased the expression of the Bax protein and cleaved caspase-3, whereas it downregulated the expression of the antiapoptotic protein Bcl-2. These changes were reversed by aliskiren. Several reports have implicated MAPK pathways in apoptotic signaling by TGF-ß. For example, activation of TGF-ß-activated kinase-1 (TAK-1), a protein of the MAP kinase family, activates p38 and JNK signaling in TGF-ß-family-induced apoptosis, 30 and p38 signaling is required for TGF-ß-induced apoptosis in murine podocytes. 31 Inhibition of TGF-ß1 and MAPK expression by aliskiren suggests that aliskiren prevents apoptosis and tubular atrophy through the inhibition of TGF- ß1 expression.
In conclusion, the renin inhibitor aliskiren exerts anti-inflammatory effects through the inhibition of NF-κB, resulting in reduction in TGF-ß1 expression and TGF-ß1-induced Smad-2/3 and MAPK signaling in GM-induced nephrotoxicity. This inhibition of TGF-ß1 signaling also attenuates GM-induced renal tubular cell apoptosis and fibrosis.
Footnotes
Funding
This work was supported by the Korea Science and Engineering Foundation through the Medical Research Center for Gene Regulation (grant number 2012-0009448) at Chonnam National University and Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (grant number KRF-20100008732), by Chonnam National University Hospital Research Institute of Clinical Medicine (grant number CRI10035-1) and by Chonnam National University (grant number 2009-2477).
Conflict of interest
None declared.